Stem Cell Engineering Developments and Advances in Providing HIV Immunity
Joseph DeSimone and Vincent S. Gallicchio*
Department of Biological Sciences, College of Science, Clemson University, Clemson, SC 29636
*Corresponding author: Vincent S. Gallicchio, Department of Biological Sciences, College of Science, Clemson University, Clemson, SC 29636
Citation: DeSimone J, Gallicchio VS. (2021 Stem Cell Engineering Developments and Advances in Providing HIV Immunity. J Stem Cell Res. 2(2):1-21.
Received: August 27, 2021 | Published: September 10, 2021
Copyright© 2021 genesis pub by DeSimone J, et al. CC BY-NC-ND 4.0 DEED. This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-No Derivatives 4.0 International License., This allows others distribute, remix, tweak, and build upon the work, even commercially, as long as they credit the authors for the original creation.
DOI: https://doi.org/10.52793/JSCR.2021.2(2)-23
Abstract
Current research in HIV treatment is oriented toward developing a therapeutic that can permanently reduce viral load and restore functional immunity but without life-long and daily drug intake. A treatment like this would ultimately provide a cure for HIV/AIDS. The transplantation of hematopoietic stem cells is one of these possible methods. These cells can be genetically engineered with HIV resistance then transplanted into a patient, where they can differentiate and mature to develop an HIV-resistant immune system. This would provide a patient with life-long resistance to HIV and reduce the effects of AIDS and further viral replication. Thus, this method of treatment would provide a one-time, life-long cure for HIV/AIDS. Here we review current and recent developments in hematopoietic stem cell modifications, anti-HIV gene construction, in vitro HIV resistance, transplantation of modified cells, and the progress of different methods on animal and human trials.
Keywords
HIV; Stem cells; Gene editing; Therapy; Immunity
Abbreviations
AAV: Adeno-associated virus; AIDS: Acquired Immunodeficiency Syndrome; ART: Antiretroviral Therapy; BCNU: Bis-chloroethylnitrosourea; BLT: Brain/Liver/Thymus; CAR: Chimeric Antigen Receptor; CCR5: C-C Chemokine Receptor Type 5; CRISPR: Clustered Regularly Interspaced Short Palindromic Repeats; CXCR4: C-X-C chemokine receptor type 4; DHR: Direct Homologous Recombination; DSB: Double Strand Break; EGFP: Enhanced Green Fluorescence Protein; FACS: Fluorescence-Activated Cell Sorting; FI: Fusion Inhibitor; GALT: Gut-Associated Lymphoid Tissue; GFP: Green Fluorescence Protein; gRNA: guide RNA; HAART: Highly Active Antiretroviral Therapy; HIV: Human Immunodeficiency Virus; HIVgp41: HIV glycoprotein 41; HSPC: Hematopoietic Stem/Progenitor Cells; INSTI: Integrase Strand Transfer Inhibitor; LTR: Long Terminal Repeat; mRNA: messenger RNA; MGMT: P140K O(6)-methylguanine-DNA-methyltransferase-P140K; NHEJ: Non-Homologous End Joining; NOG mice: NOD/Shi-scid/IL-2Rγnull mice; NNRTI: Non-Nucleoside Reverse Transcriptase Inhibitor; NRG mice: NOD-RAG1−/− IL2rγ−/− mice; NRTI: Nucleoside/Nucleotide Reverse Transcriptase Inhibitors; NSG mice: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice; OZ1: anti-HIV ribozyme targeting tat/tar; PBMC: peripheral blood mononuclear cell; PCR: Polymerase Chain Reaction; PKC:Protein Kinase C; RNAi: RNA interference; RISC: RNA Induced Silencing Complex; shRNA: short hairpin RNA; siRNA: Small Interfering RNA; TALEN:Transcription Activator-Like Effector Nuclease; TAR: Trans-acting Response element; ZFN: Zinc Finger Nuclease
Introduction
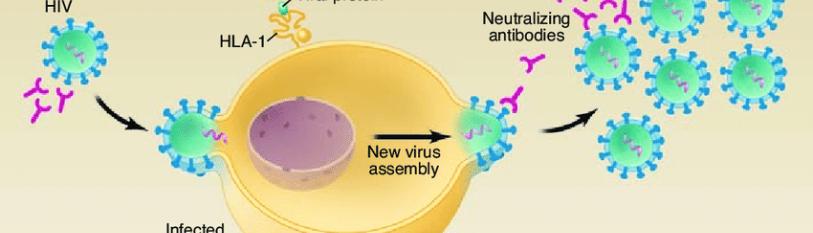
Human Immunodeficiency Virus (HIV) is a virus that attacks the body’s immune system by targeting its cells, integrating its genome into the host cell, then expressing the proviral genome and constructing mature viruses that bud from the cell [1]. HIV virus is able to target these cells by specifically binding to CD4 protein receptors on the surface of the cell membrane that normally function in immune recognition. The virion acquires particles of the cell membrane including certain spike proteins and the lipid envelope [2], leaving the previously infected immune cell without a protective membrane. This immune-targeting nature of HIV is what makes the virus so dangerous. CD4+ cells are crucial to the immune system and as virus maturation takes place, the body’s defense to other infections reduces drastically. Normal CD4+ cell counts, also referred to as white blood cell counts, range from 500-1,200 cells per cubic millimeter of blood, whereas HIV-infected patients range between 30 and 1,000 cells per cubic microliter of blood; this is 109 reduction in white blood cells [3].
With an almost non-existent immune system, HIV positive patients can develop Acquired Immunodeficiency Syndrome (AIDS). AIDS is clinically indicated by a reduction in white blood cell counts, high levels of HIV antibodies in the blood (referred to as viral load), and the development of additional diseases [4]. As of June 2021, over 75 million people have been infected with HIV/AIDS and 32 million have died world-wide since the first reported case on June 5, 1981 [4].
In 1996, highly active antiretroviral therapy (HAART) was introduced to medical practice to treat HIV-infected individuals [5]. HAART involved the administration of several drugs that inhibit several mechanisms of viral replication to prevent propagation and emergence of resistant strains [5]. The specific drug cocktail can differ between the pharmaceutical companies that produce it, but there are 6 main classes of drugs involved that target the virus lifecycle: NRTIs, NNRTIs, PIs, INSTIs, FIs, and CCR5 Antagonists [5]. Nucleoside/Nucleotide Reverse Transcriptase Inhibitors (NRTIs) include drugs like abacavir, lamivudine, tenofovir, and zidovudine. They are nucleotide analogs which competitively bind to the reverse transcriptase enzyme and prevent the growth of viral DNA upon infection [5]. Non-nucleoside Reverse Transcriptase Inhibitors (NNRTIs) include drugs like delavirdine and nevirapine which also bind to HIV reverse transcriptase and inhibit viral DNA growth, but they are not nucleotide analogs. Instead, they bind to a hydrophobic, allosteric region of the enzyme [5]. Protease Inhibitors (PIs) include drugs like atazanavir and indinavir which inhibit the cleavage of gag/pol polyproteins and prevent maturation of the virion and cause it to become non-infectious. Integrase Strand Transfer Inhibitors (INSTIs) include drugs like dolutegravir and raltegravir and prevent HIV DNA from incorporating into the host cell DNA by binding HIV integrase enzyme [5]. Fusion Inhibitors (FIs), like enfuvirtide, prevent HIV from fusing with CD4+ cells by binding to the glycoprotein gp41. Finally, Chemokine Receptor Antagonists (CCR5 antagonists) prevent viral entry by inhibiting the interaction between the viral envelope glycoprotein gp120 and CD4+ cells and include the drug maraviroc, to name one [5].
HAART proved to be extremely effective in suppressing HIV viral load to undetectable levels, but daily drug treatment is required and after a few weeks without drug intake, viral load rebounds in a majority of the patients, indicated by a reduction in levels of HIV antibodies [6]. However, 14 patients interrupted HAART therapy, which was given in the early stages of infection, and they showed maintenance of low viral loads for several years [7]. This is a unique scenario because viral load has been shown to rebound when HAART is interrupted. Most patients require daily life-long drug administration to prevent HIV replication and maintain healthy white blood cell counts. This can be a difficult protocol to follow and is an expensive therapy, and there have been several side effects reported as HAART accumulates over time.
There have been reports of cardiovascular diseases and early signs of senile complication with dyslipidemia in patients that suffer from chronic HIV infection, even when receiving HAART [8-10]. These symptoms are common indicators of AIDS, one of the things HAART aims to prevent. Therefore, a novel therapeutic strategy that provides lifelong remission of HIV/AIDS, or a cure to HIV, is a major focus in HIV research.
The first and only case of a patient cured of HIV was reported in a CCR5-deficient bone marrow transplant in 2009 [11]. Timothy Brown, also known as the Berlin Patient, developed Acute Myelogenous Leukemia (AML). He also was HIV positive, so he was able to be enrolled in a trial to cure his leukemia. Naturally HIV resistant CCR5 homozygous D32/D32 stem cells found in bone marrow were transplanted and fully reconstituted his immune system through these resistant cells. CCR5 is one of the chemokine receptors that facilitates viral fusion to the cell membrane, and some individuals have a natural mutation (D32/D32) where their cells do not express this receptor. Thus, they have a resistance to HIV and these resistant, transplanted cells reconstituted the Berlin Patient’s immune system. HIV viral load was undetectable for the rest of his life until leukemia returned, and he died in hospice on September 29, 2020. [11]. The success of this case created excitement and interest in finding a cure to HIV through a similar method, but of the several attempts to repeat this success, none were able to achieve HIV-curing results [12-15]. For example, a few HIV positive patients have the CCR5 deletion which is explained by the HIV population in their body. It contains a strand of HIV (X4-tropic HIV) that uses CXCR4, an alternative chemokine receptor [12]. A study by Henrich et. al found undetectable levels of HIV-1 in peripheral blood after HSPC transplantation [15]. However, after 12 and 32 weeks, the 2 patients in the study experienced viral rebound [15]. They speculate that long-lived tissues may serve as viral reservoirs that can reemerge over time.
More current research to find a cure for HIV/AIDS has shifted focus from transplants of naturally resistant donors to using genetic engineering to develop patient-derived hematopoietic stem/progenitor cells (HSPC) that are modified to prevent HIV infection. These transplantable stem cells could, in theory, be feasibly re-introduced to the patient and reconstitute the immune system with HIV. This method of stem cell engineering would not only provide HIV resistance but could also be a cure to HIV that possibly would require only requiring one treatment.
Methods of Engineering HIV Resistance in Stem Cells
A method of engineering HIV immunity in stem cells is centered on the manipulation of the chemokine receptor CCR5 [16]. After HIV binds to the main CD4 receptor on the cell’s surface, it subsequently binds to CCR5 and can then enter the cell [16]. Thus, by preventing the expression of CCR5, HIV infection can be avoided by preventing entry into the cell and ultimately integration into the genome. Fortunately, lack of CCR5 expression is not detrimental to general immune system functions and hematopoietic effects [17]. Individuals with homozygous D32/D32 mutations in the CCR5 gene do not express this receptor, but some have been found to be more susceptible to West Nile virus [18]. Others with this mutation reported a reduction in portal inflammation and milder fibrosis after hepatitis C infection [19-21]. Most importantly, the homozygous CCR5 D32/D32 mutation creates a natural resistance to HIV transmission, and even those that are heterozygous for the mutation have shown a slower disease progression to AIDS by about 2-3 years when compared to individuals with wild type CCR5 gene expression [21].
CCR5 knock out via CRISPR/Cas9
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) is a gene editing technology derived from the bacterial adaptive immune system [22, 23] and has been used to knock out the CCR5 gene [24]. A double strand break (DSB) is created in the DNA by the Cas9 protein which is directed by a specific guide RNA (gRNA) sequence. These gRNAs can be customized to direct the Cas9 protein to cleave virtually any sequence, in this case the CCR5 gene [24]. The first group to successfully develop CCR5 inhibition via CRISPR/Cas 9 were able to induce a 33% reduction in cleavage and homozygous mutation in HEK 293T cells [24]. A year later, Mandal et. al achieved similar levels of mutation (20-40%) of the CCR5 gene in K562 cells and CD34+ HSPCs [25]. They also achieved a biallelic mutation of CCR5 in 26% of the CD34+ HSPCs by using a dual gRNA strategy [25]. This result in a biallelic mutation is the end goal of CCR5 knock out, and this method is under further development for optimization.
Ribozyme Inhibition of CCR5 mRNA
Ribozymes, RNA molecules that can function as enzymes and cleave other RNA targets, have been designed and manipulated to target CCR5 mRNA sequences [26,27]. Cagnon et. al was able to construct a ribozyme from a modified Adenovirus VA1 ribozyme that targets CCR5 mRNA [28]. The PM1 cell line that was transduced with this ribozyme had a 70% reduction in CCR5 expression levels and was resistant to R5-tropic HIV, but did not provide resistance to R4-tropic HIV, an emergent strand of HIV-1 that does not require CCR5 for fusion but can instead use CXCR4 [28]. Li et. al used this ribozyme in a combinatorial anti-HIV technique that also involved the use of a short hairpin RNA (shRNA) which targets HIV tat and can serve as a TAR decoy [29]. 96-98% of T293 cells were effectively transduced with these vectors, and 15-20% of peripheral blood mononuclear cells (PBMCs) were transduced. This ultimately led to an increase in HIV resistance in CD34+ HSPCs indicated by a reduction in HIV-1 p24 antigen [29].
HIV Resistance in Hematopoietic Stem Cells
An obstacle in developing HIV-resistant stem cells is that the virus can infect HSPCs in the patient [30, 31]. This is problematic because attempting to reconstitute a patient’s immune system requires the use of HSPCs. However, it has been found that HSPCs expressing the CD34 receptor are immune to HIV infection [31]. Isolated CD34+ HSPCs have been reported to resist HIV-1 infection in-vitro; some subpopulations of these cells express CD4 receptors in various degree, and others even express CXCR4 receptors at high frequencies [31]. Another study by Shen et. al isolated CD34+ from normal adults to examine their interactions with HIV-1 [32]. These cells also expressed CCR5, CXCR4, and CD4 receptors, but the levels of expression of these receptors was insufficient for viral infection [32]. Thus, stem cell resistance to HIV exists, and isolation of specific populations of cells is required to not only study techniques of gene-editing but to also reconstitute an infected patient with resistant stem cells.
RNA Interference Through sh1005
Another technology involved in specified mRNA degradation is RNA interference (RNAi) via the RNA Induced Silencing Complex (RISC) [33]. This technology has been used to reduce CCR5 expression. A method developed in 2003 by Qin et. al involved the use of shRNA that was delivered through a lentiviral vector and targeted the CCR5 gene [34]. CD4+ T cells that were transduced saw a 10-fold decrease in CCR5 expression and were 3 to 7 times more resistant to R5-tropic HIV [34]. This shRNA used a type of polymerase promoter called U6 RNA polymerase promoter to knock down CCR5 sufficiently, but this promoter expressed large amounts of shRNA that were cytotoxic to the cells. However, it was found that non-toxic expression could be achieved while maintaining sufficient levels of shRNA expression by using a slightly weaker H1 RNA polymerase III promoter instead [35]. An et. al was able to develop a different shRNA that targeted CCR5 and was tested on human primary T-cells [36] and CD34+ cells in vitro [37]. This new shRNA, named sh1005, was able to efficiently modify the cells and reduce CCR5 mRNA levels (>90%) and induced some resistance to R5-tropic HIV [37]. sh1005 was then tested in other studies and was found to down regulate CD4+ T cells in lymphoid tissues of humanized mice models [38], increase resistance to R5 tropic HIV after challenge in humanized mice [39].
Zinc Finger Nucleases
A Zinc Finger Nuclease (ZFN) is a DNA cleavage domain that is derived from the restriction endonuclease FokI [40]. It is fused to a zinc finger DNA binding domain which, when specifically directed, can target and disrupt genomic DNA creating a double strand break (DSB) [40]. The first successful in vitro targeting of CCR5 via ZFN was published in 2005 [30]. Essentially, the Zinc Finger domain targeting the CCR5 regions binds it, then the restriction endonuclease FokI induces a DSB at the site, and the non-homologous end joining (NHEJ) system repairs the cleavage site [41]. This technique was later tested and published in another study by Perez et. al that measured the efficiency of the method on primary human CD4+ T cells and found that expression of CCR5 was reduced by around 50% [42]. Further, the cell lines with reduced CCR5 expression were stable and protected from infection upon HIV-1 challenge in vitro [42]. Later, human CD34+ HSPCs derived from umbilical cord blood and fetal liver yielded a 17% disruption in CCR5 alleles via ZFN technique [42].
The DSB capabilities of ZFN can also be used to insert anti-HIV genes at the CCR5 gene through a direct homologous recombination (DHR) [43]. DHR is a mechanism that cells utilize following a DSB in which the homologous nucleotide sequence serves as a template for DNA repair, and this method has been successfully utilized to insert genes into a target region [43]. This method allows genetic engineers to provide a template containing a desired sequence to integrate into a locus region that was removed; specifically, anti-HIV genes can be inserted into the CCR5 locus. Wang et. al successfully electroporated a ZFN AAV stereotype 6 vector containing a donor template into mobilized peripheral blood CD34+ HSPCs. A GFP cassette was inserted at the CCR5 with a 17% success rate [44]. This technique was also applied to fetal liver HSPCs in the study with a 19% success rate, and these cell lines differentiated into modified CD34+, CD133+, and CD90+ cells which matured into HSPCs. The study also found that the amount of DNA template correlated with the mutation frequency and GFP expression.
CCR5 knock out via TALEN
Similar to ZFN knock out, transcription activator-like effector nucleases (TALENs) can be used to target regions of the genome and induce a cleavage. TALENs also contain the Fok1 nuclease, but it is fused to a DNA binding domain of transcription activator-like effectors. The advantage to TALENs is that the DNA recognition sequence is between 33 and 35 nucleotides (versus ZFNs that range from 18 to 36 nucleotides) [43-46]. This technique was proven effective on the CCR5 region by a group in 2011 that compared knock rates to ZFN. HEK293T cells were transfected with a vector containing CCR5-targeting TALEN and ZFN. They found that 17% of the cells were modified by TALEN, versus the 14% modified in the control group transfected with ZFN [46]. Later, another group developed a more specific guiding sequence for TALEN and transfected it into primary human T-cells. They found that this design caused CCR5 knock out by more than 50%, which showed resistance to R5 tropic HIV upon challenge in vitro. A more recent study transfected primary CD4+ cells and achieved 89% disruption of the CCR5 gene [47]. They then compared the edited cells to a control group and found no significant differences in proliferation nor cytokine secretion capabilities.
Accuracy and Modification of CRISPR vs TALEN
A common issue in genome editing in any field is the chance of inducing unintended effects, and different techniques have shown different levels of off-target cleavages. Between the three previously discussed knock out techniques, TALENs are the most specific and less frequently induce non-target changes [48]. However, CRISPR/Cas9 targeting CCR5 had significant effects in other regions, specifically at the locus for CCR2, another gene coding a different chemokine receptor that is similar in sequence to CCR5. In a study by Cradick et. al measuring the off-targets effect of this technique, five different gRNAs targeted to CCR5 were used on CRISPR/Cas9 to compare efficiencies, and they found that, on average, 57% of the CCR5 genes were successfully mutated [49]. They also measured unintended CCR2 mutations and found that one gRNA induced mutations at the locus as low as 5%. There was still a need to optimize the technology and reduce off-target mutations, and a study by Yi et. al focused on comparing its specificity to that of TALEN. The group found gRNA sequences unique to the CCR5 region and found that, when using these gRNAs, 4 times the number of CRISPR/Cas9-treated cells underwent editing compared to those treated with TALEN [50]. Thus, CRISPR/Cas9 is more efficient in knock-out, but not as accurate as TALEN. Another notable result from this study is that mutations occurred in both alleles 33% of the time using the CRISPR/Cas9 method, whereas TALEN only edited both alleles at 10% frequency. In general, the goal of reducing off-target mutations is a method of optimizing CRISPR/Cas9, and Kleinstiver et. al developed a mutated Cas9 protein that can reduce these undesired mutations. They used information on the protein’s structure to make 4 amino acid substitutions that increased its DNA and gRNA binding affinity [51]. This adjusted Cas9 protein proved to operate at the same efficiency and off-target mutation levels were undetectable. These adjustments to the protein imply a promising future for the specific technology.
CXCR4 Inhibition Inhibits X4-Tropic HIV Infection
CCR5 is an essential coreceptor for HIV entry, but studies have shown that a different strain of the virus, specifically X4 tropic HIV, utilizes another coreceptor for entry: CXCR4 [52]. Thus, it is critical to develop a safe method of engineering CXCR4 inhibition because an X4 tropic HIV strand could emerge in a R5 tropic-selective setting. In fact, this occurred in a recent clinical case in which the patient was transplanted with HSPC cells from a donor that was D32/D32 homozygous [14]. The cells did not express CCR5, so HIV inhibition was expected, but there was also a rapid rise in X4 tropic HIV due to the availability of alternative coreceptors. Since, there have been some efforts to develop a mechanism to downregulate CXCR4 expression [52]. Anderson et. al in as early as 2003 developed an shRNA that targeted and inhibited CXCR4 expression [53]. Downregulation was confirmed via Western Blot analysis, and the treated cells persisted after X4 tropic HIV challenge. The previously discussed genome editing technologies have also been used, like in one study by Yuan et. al that utilized a ZFN to knockout CXCR4 in primary T cells and a CD4+ T cell line [54]. The cell lines were stable and resisted HIV challenge. Another study focused on the capabilities of CRISPR/Cas9 to knock out CXCR4. They found that the technology could knock out the gene in primary CD4+ lymphocytes with a success rate ranging from 18 to 22% [55]. It is important to note that although this gene target has potential to provide HIV resistance, it has only been successfully downregulated in CD4+ T cells. This receptor has been known to play a role in cell proliferation, communication, migration, and other signaling pathways so it may not be a viable target for HSPCs [56]. Furthermore, when CXCR4 was knocked out in both alleles of in utero mice, it was found to be lethal [57], or produced embryos with bone marrow deficiencies and immune dysfunctions. Thus, this strategy may not be an effective method of altering HSPCs but could be used to engineer HIV resistance in mature CD4+ cells. This method of HIV resistance is more of a preventative measure than a main focus of current research, but still stands to be an important aspect of stem-cell based HIV immunity.
Preventing Viral Entry via C46 Fusion Inhibitor
C46, an anti-HIV gene, has also been studied and used to prevent HIV entry [57]. The protein expressed is anchored in the membrane of the cell and expressed on the surface, where it interacts with HIV. The N-terminal alpha helix of HIV glycoprotein 41 (HIVgp41) interacts with the C46 gene protein. This interaction interferes with the HIV six-helix bundle and ultimately prevents fusion to the cell membrane, providing HIV resistance to the cell [57,58]. This C46 inhibition has proven to be effective in preventing both R5 tropic and X4 tropic HIV strands in vitro and in humanized mice [59]. A clinical trial was later conducted by van Lunzen et. al in which a retrovirus containing the C46 gene was transfected into autologous T cells, which successfully survived HIV challenge [60]. The cells were introduced to HIV-positive individuals with no significant adverse effects were reported in any of the seven patients. No gene marking levels were increased. However, there was no observed decrease in viral load in any of the patients [60]. However, these results provide grounds for improvement on an effective genetic modification and reintroduction method. This method has also been used by Wolstein et. al in combination with shRNA knockout of the CCR5 gene in CD4+ T lymphocytes and CD34+ HSPCs [61]. This method was successful in delivery to the cells in vitro and the cells survived R5 and X4 tropic HIV challenge [50].
TRIM5alpha/TRIMCyp Host Restriction Factors
Another HIV prevention mechanism that has been studied is that of host restriction factors, an innate (or in some cases adaptive) antiviral system in host cells that respond to viral infection [62]. Typically, these proteins are expressed in low levels, but upon response to viral infection, their expression is upregulated in response [62]. These proteins provide a first line of defense against infection, but HIV can escape these host restrictions and effectively infect host cells. Interestingly, other species have host restriction factors that can provide an effective antiviral defense against HIV. One study by Chan et. al found that the Rhesus macaque TRIM5alpha, a restriction factor also found in humans, was effective in preventing HIV infection [62]. The human version of this restriction factor cannot, but the Rhesus macaque version binds to the HIV proteins and directs them to a proteosome for degradation. Unfortunately, the Rhesus macaque TRIM5alpha protein has been found to induce immunogenicity, so it cannot be used as in clinical settings [62]. Yap et. al further investigated a safe modification to human TRIM5alpha and found that inducing a single amino acid substitution (R332P) enabled the TRIM5alpha to bind HIV capsid proteins and restrict viral infection [63]. This can partially be explained by minor evolutionary changes to genomes between humans and Rhesus macaque. After this discover, Neagu et. al found that the species douroucoulis (or owl monkey) have a TRIMCyp fusion protein that is an effective defense against HIV [64]. They genetically engineered a human version of this host restriction factor, HuTRIMCyp. The modified gene was integrated into a lentiviral vector and introduced to primary CD4+ T cells and macrophages and found that it was able to bind HIV capsid and prevent infection after the virus enters the cell [64]. Immunodeficient mice were introduced to these modified human CD4+ T lymphocytes and showed a decrease in viral load [64]. These results are evidence that TRIMCyp and TRIM5alpha can be an effective method of genetically engineering HIV resistance.
Post-Infection HIV Inhibition
The previously mentioned techniques provide HIV resistance by preventing infection and integration into the host genome. These methods are critical in engineering complete HIV immunity, but it is equally important to focus on systems that prevent HIV replication in the host in case of infection. One strategy targets provirus integrated into the host genome. In 2007, Karpinski et. al modified a Cre recombinase to target the 5’ end long terminal repeat (LTR) of HIV by modifying its specificity [65]. This new recombinase (Brec1) can recognize a 34-base pair LTR of HIV provirus [65]. Brec1 was introduced to an HIV provirus derived from a patient and successfully excised the HIV genome [65].
Another strategy involves ZFNs. Qu et. al infected a line of Jurkat T cells with an HIV strain containing EGFP gene to serve as a marker [66]. Infected cells were selected and, via nucleofection, were introduced to a modified ZFN that targets the LTR in HIV. After three days the genome of the infected cells was analyzed and confirmed that provirus was excised [66]. This technique was also used on a line of primary T cells and found similar results [66]. Ebina et. al used CRISPR/Cas9 to target latent provirus using gRNAs that recognize the LTR of HIV and reduced levels of provirus in Jurkat cells by 30% [67]. However, the NHEJ abilities of this technology has been shown to create viral escape variants when nonessential viral sequences were targeted [68].
Another anti-HIV technique that prevents replication after infection revolves around HIV Tat and TAR [69]. These two factors interact with each other and promote the expression of the viral genome through RNA transcripts [69]. Lee et. al found that HIV transcription was inhibited when they introduced a small interfering RNA (siRNA) that targeted Tat in infected macrophages, and later demonstrated similar results using shRNA [70]. Strong et. al successfully developed a TAR-targeting TALEN that could disrupt the sequence in the provirus [71]. A HeLa cell line was modified to express EGFP, then was treated with the specific TALEN, and they saw a 55% reduction in EGFP expression in the cells, and that 20% of the provirus was disrupted in the HeLa cell line [71].
Combinatorial Anti-HIV Gene Editing
An important facet of HAART is its ability to target multiple mechanisms required for HIV replication [8]. This success in multi-target treatment suggests that anti-HIV genetic engineering might need to use a combination of techniques to see similar success. Combinatorial techniques would specifically aim to prevent the emergence of resistant HIV mutants. For example, a CCR5 D32/D32 donor provided HSPCs that were transplanted to a patient which resulted in a CXCR4 tropic HIV strain emergence [8]. ter Brake et. al demonstrated this by using multiple shRNA treatments on a line of CD4+ cells [72]. The cells treated with shRNA that targeted only one protein-coding region (Gag-5, Pol-1, Pol-47, or R/T-5) saw a slight increase in HIV resistance when compared to the control, but HIV escape mutants were detected [72]. Most importantly, the CD4+ cells transfected with all four shRNAs showed the greatest resistance and no mutant strains emerged [72]. One strategy by Walker et. al successfully developed a method in vitro and eventually tested in humanized mice [73]. This group combined a CCR5-targeting shRNA, a TAR decoy, and TRIM5alpha derived from rhesus macaque. This combination of anti-HIV genes was inserted into a lentivirus and transduced into CD34+ HSPC. The cells were successfully engrafted into mice. Another combination strategy by Anderson et. al used 3 anti-HIV genes, specifically an shRNA targeting rev and tat mRNA, a TAR decoy that would prevent the binding of the transactivation response to tat, and a CCR5 ribozyme. These genes were inserted into a lentiviral vector. Their construct, “Triple-R”, was transfected into CD34 macrophages and a line of T cells, and resisted HIV infection in vitro without any escape mutants [74]. This method was replicated by DiGiusto et. al in a phase I clinical trial where CD34+ progenitor cells were treated with Triple-R. Gene marking was as high as 22% in vitro and upon transplant into 4 patients; there were no adverse effects [75]. Throughout the 24-month observation, vector marking was persistent in cell lineages. Although the levels were low, this demonstrated the capabilities of CD34+ HSPC modification and transplantation.
The results of these studies not only proved that a combination of anti-HIV genes was optimal, but that it could also be effectively used on stem cells without major deficiencies in growth or maturation. It also helped protect against HIV infection, and the successful engraftment of modified HSPCs revealed that a treatment was very possible. In fact, it is common to see others build on the success of another group to further optimize the method. For example, a paper by Ringpis et. al was published in 2012 that highlighted the success of an shRNA targeting LTR of HIV combined with sh1005 [76]. Together, there was an almost 7-fold reduction in HIV replication [76], and the CD34+ HSPCs that were transduced differentiated in vitro and PBMCs that developed maintained the shRNA expressions. This stable differentiation showed promise in a double-shRNA combination, and in 2014 a paper by Wolstein et. al was published that built off that success [77]. They combined sh1005 with C46 inhibitor RNA and transfected the two into CD34+ HSPCs via lentivirus. The PBMCs showed resistance to both R5 tropic and X4 tropic HIV strands [77], which was later engrafted into BLT humanized mice. The 2015 paper by Burke et. al that detailed the results of the BLT mouse engraftment, which followed the previously mentioned study, showed that the mice supported multilineage hematopoietic reconstitution and HIV viral load did not significantly increase upon challenge [48].
Anti-HIV Genes In Vivo
With great success in developing methods to engineer HIV resistance in stem cells, many studies continue to the next stage of investigation of the treatment as an ultimate therapy for HIV. By using an animal models, research teams can get a better understanding of the implications of engraftment, hematopoietic reconstitution, and in vivo immunity. Humanized mice are mice that have been xenotransplanted with human cells or have been modified to express human gene products. These mouse models serve as good initial indicators of how certain methods will reconstitute and perform in humans and major issues can be recognized and fixed during this step [78]. One of the first few studies to find some success in a therapy was published in 2010 by Holt et. al [79]. After effectively delivering CCR5-targeting ZFNs to CD34+ HSPCs, the team transplanted the modified cells into NOD. Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice [79] (NSG humanized mice can support human hematopoiesis and are subject to HIV infection). The modified CD34+ HSPCs were successfully transplanted and multilineage differentiation was observed indicated by a CCR5 disruption between 5 and 7% in the NSG mice. Secondary transplantation was performed on the mice and CCR5 disruption increased (12-20%) and the mice were infected with R5 tropic HIV. This served as a method of selection for cells that did not express CCR5, and actually led to a decrease in viral load over time and human T-cell populations increased 12 days after infection [79]. Saydaminova et. al repeated the same technique in CD34+ HSPCs and CCR5 disruption was as high as 12% upon transplantation into NOD/Shi-scid/IL-2Rγnull (NOG) mice [80]. One issue this study had was the survival of the CD34+ cells. When compared to HSPCs that were not transduced by the CCR5-targeting ZFN, the modified cells had lower levels of engraftment and differentiation. This negative effect could potentially come from the extended expression of ZFN in the cells, which has been proven to be cytotoxic [80].
This is important to consider in understanding the effects of certain DNA-modifying techniques. A more recent preclinical study by Hofer et. al focused on these cytotoxic effects in humanized mice [81]. They studied the effects of ZFN disruption of the CCR5 gene in mobilized peripheral blood derived HSPCs and focused on intravenous injection into adult NSG mice. This treatment resulted in engraftment levels near 100% which was maintained for more than 6 months and found that treating HSPCs with protein kinase C (PKC) activators before ZFN transfection led to over 25% disruption of CCR5 [81]. Another study by Li et. al attempted to optimize ZFN disruption of CCR5 was published in 2013 [82]. HSPCs were isolated from granulocyte colony-stimulating factor-mobilized adult blood and treated them with a recombinant adenoviral vector of CCR5-targeting ZFN [82]. They also found that pretreating HSPCs with PKC activators promoted ZFN disruption and adenoviral protein expression; over 99% of the cells were modified and CCR5 gene disruption was greater than 25%. They also demonstrated that by optimizing adenoviral vector PKC activator levels, the modified HSPCs could be successfully engrafted into NSG mice models [82]. The level of gene-modified cell differentiation was low when compared to gene marking in the untreated group, but it still demonstrated the ability to support in vivo differentiation.
A study in 2005 conducted by Li et. al produced a combinatorial vector containing tat/rev siRNA, a ribozyme targeting CCR5, and TAR decoy that effectively suppressed HIV infection in HSPCs long term [83]. This treatment of HSPCs progressed to a plot clinical trial in which four patients diagnosed with AIDS were already receiving HSPC transplantation as a lymphoma therapy. Some HSPCs were transfected with this combinatorial vector and gene-modified cells engrafted without adverse effects, multilineage transgene expression was observed and provided evidence that this method of treatment was possible in patients [83]. Unfortunately, peripheral blood mononuclear cells were measured for gene marking, and the frequency was too low (0.02-0.32%) to measure HIV resistance. A study published in 2014 by Chung et. al tackled this challenge of increasing frequency by developing an O(6)-methylguanine-DNA-methyltransferase-P140K mutant (MGMT-P140K) drug resistance marker into a vector containing three antiviral RNAs derived from MCM7 gene [6]. HSPCs were transfected with this combinatorial vector and MGMT-P140K marker and were transplanted into immunodeficient NSG mice bone marrow and spleen. They were then treated with bischloroethylnitrosourea (BCNU) and O(6)-benzylguanine for selection, which resulted in up to a 15 fold frequency increase in CD45+ cells when compared to the immunodeficient control group. This shows that selective enrichment occurred in-vivo and can be used on HIV-target cells [6].
Another 2015 study by Myburgh et. al focused on the genetic modification of CD34+ HSPCs via lentiviral vector to downregulate CCR5 and its effectiveness in providing HIV resistance in vitro and in vivo [84]. Using FACS sorting, the group separated CD34+ HSPCs into those that received effective CCR5 knockdown (FACS-sorted R5 knockdown) and transplanted the cells into NSG mice. This resulted in lower viral loads in the FACS-sorted R5 knockdown mice when compared to those treated with FACS-sorted negative HSPCs and the control mice [85]. Over the course of 28 weeks, they also noticed that the FACS-sorted negative mice saw a decrease in CD4+ T cells (from 55% to 20% in 134 days), while the FACS-sorted R5 knockdown mice saw a steady increase in CD4+ T cells (from 33% to 65% in 196 days). These results show that transplantation of modified HSPCs with high levels of CCR5 knockdown can induce lasting HIV inhibition in vivo and promote an increase in resistant target cells. Wang et. al, who were successful in developing a CCR5-targeting ZFN, transfected mice with HSPCs that received CCR5 knockout [44]. Fetal liver CD34+ cells were transduced with a CCR5-GFP AAV6 to measure CCR5 expression levels, then transfected the cells with CCR5 ZFN mRNA [44]. These modified cells matured and were engrafted into neonatal NSG mice. Throughout the 16-week study, they analyzed peripheral blood bone marrow and spleen and detected an increase in levels of human CD45+ when compared to the control mice and reported that the modified CD34+ cells underwent multilineage differentiation [44]. They also used flow cytometry on the treated mice cells to measure genome editing in cells found in blood and tissues. They found several lineages, including the CD45+ cells, expressed levels of genome editing, and in-out PCR was conducted on these cells which confirmed GFP insertion on the CCR5 gene in the treated group. This revealed the success of their genome-editing technique and proves that differentiation of modified HIV-resistant stem cells can occur in vivo. Finally, they harvested the modified HSPC-rich bone marrow of the treated mice and performed a secondary transplant on another group of NSG mice. After 20 weeks, they measured gene-editing frequency in the HSPCs and reported similar or higher levels of modification when compared to the original NSG mice donors [44]. This conclusion is incredibly significant because it reveals that these modified cells can not only be successfully transplanted between organisms, but they can differentiate and increase genome editing in vivo.
Another previously mentioned combinatorial technique combines multiple shRNAs into a single lentivirus, which found that expression increases when each shRNA is driven by a different promoter [72]. In the 2008 study by ter Brake et. al, a method was developed to measure the potency of anti-HIV gene levels in humanized mice, which is important to evaluate the safety of this specific gene modification. Later, in 2013, Centrlivre et. al used a similar combination technique of 4 anti-HIV shRNAs which targeted the tat/rev open reading frames, viral capsid, protease, and integrase for all classes of HIV [85]. They individually tested the effects of each shRNA, and found that one of the four, which targets the Gag gene, negatively impacted development of the hematopoietic cells in vivo. However, shRNAs targeting tat/rev, CCR5 ribozyme, and TAR decoy were found to be safe and did not affect multilineage differentiation in vivo. These 3 shRNAs were combined into a single lentiviral vector and were transfected to several HSPCs which were transplanted into humanized mice which resulted in multilineage differentiation in vivo, and CD4+ T cells were harvested and found to resist HIV upon challenge [85]. These results provide some evidence that shRNA combination treatment can not only provide resistance to HIV and can also prevent the emergence of escape mutants.
Another combinatorial technique was published in 2012 by Walker et. al that evaluated in vitro and in vivo efficacy of the treatment [73]. The developed vector combined a CCR5 shRNA, TAR decoy, and TRIM5 isoform [73]. CD34+ HSPCs were transfected with the combinatorial lentivirus then introduced them into NOD-RAG1−/− IL2rγ−/− knockout (NRG) mice and reported a 17.5% engraftment and normal multilineage differentiation in several lymphoid organs and peripheral blood of the NRG mice. The transfected NRG mice were then separated into two groups and given either R5 tropic HIV challenge, or X4 challenge. In both groups CD4+ cell levels were stabilized and maintained which can be explained by the selection of modified cells with HIV resistance [73]. By essentially creating an environment that only favors modified hematopoietic cells, the group was able to select for resistant cells and ultimately re-program HIV-target cells throughout the mice and provide a renewed and resistant immune system. This concept of HIV resistance and selection is important in understanding how a gene-modified immune system in HIV-positive patients will be reconstituted. Selection could also be important before transplantation because introducing a more cells that have anti-HIV genes can exponentially increase HIV resistance. In fact, a study published in 2012 by Kalomoiris et. al focused on selection for modified HSPCs using human CD25, a low affinity IL-2 receptor subunit that is not normally expressed on or within HSPCs and does not affect signaling pathways. This CD25 was incorporated into an anti-HIV vector which was transfected into HSPCs. Properly transfected cells expressed the CD25 marker and allowed for selection of cells that received the anti-HIV genes. After HIV challenge, the group was able to collect a population of cells that exhibited potent levels of HIV resistance (94.2%), which proves that this method can be used to purify population before transplantation [86].
Some humanized mouse models are specifically designed to mirror human marrow liver and thymus, important organs in HIV replication. These marrow (or Blood)/Liver/Thymus (BLT) mice models are good for understanding engraftment of modified stem cells, but they are specifically helpful in understanding how modified HSPC reconstitution will occur before treating human patients [87]. BLT mice are co-transplanted with human fetal thymus and liver and creates a thymus-like organoid that supports human T lymphocyte reconstitution, and the immune cells in BLT can be infected with HIV. In summary, they serve as another good model to follow the effects of HSPC modification and engraftment. Shimizu et. al, who were involved in optimizing sh1005, found success with BLT mice by using the CCR5-targeting shRNA, that was transfected into fetal liver-derived CD34+ HSPCs which were engrafted into BLT mice [28]. They observed effective knockdown of CCR5 [38], and CD4+ T cells showed in vivo resistance to R5 tropic HIV without any X4 tropic HIV emergence [39]. Ringpis et. al used the same sh1005 vector and combined it with an shRNA that targets the HIV LTR and transfected CD34+ HSPCs with both [66]. These cells were transplanted to BLT mice and measured reconstitution over 12 weeks. They measured gene modification and found some mice displayed levels as high as 80% of modification to CD45+ cells, which showed resistance to both R5 and X4 tropic HIV. These modified cells were found in spleen, GALT, and bone marrow which provides evidence that the transfected HSPCs can differentiate into human T cells in lymphoid organs and blood. Furthermore, various mouse tissues showed up to a 7.5% reduction in CCR5 expression [66]. A later study utilizing a dual vector transfected human fetal liver-derived CD34+ HSPCs with a lentiviral vector containing an C46 gene and an shRNA targeting CCR5 (LVsh5/C46) called Cal-1. The transfected HSPCs were transplanted to BLT mice which displayed levels of engraftment up to 78% and levels of modified CD4+ T-cell in peripheral blood and lymphoid organs that suggest successful multilineage hematopoietic development in vivo [59]. The modified cells maintained consistent levels over 6 months, and these results demonstrate the efficacy of Cal-1 as another modification technique.
Chimeric antigen receptors (CARs) are a recently used technology to often treat cancers. T cells are engineered to display artificial chimeric receptors that utilize antigen-binding and T cell activation to recognize and target cancer cells to destroy them more efficiently. This technology has been developed as a potential technique to modify HIV resistance in cells [88]. CD4zCAR is a is a molecule that contains transmembrane and extracellular domains of the CD4 molecule and is fused to CD3 complex z chain, a signaling domain involved in cytotoxic and helper T cell activation [88]. When HIV gp120 envelope protein is displayed on HIV-infected cells, CD4 recognizes it and the CD4zCAR cells are activated [88]. These cells have been reported to inhibit viral replication in the cell and are known to kill other infected cells in BLT mice. A study by Zhen et. al combined CCR5-targeting shRNA with CD4zCAR and demonstrated that the T cells could respond to HIV antigens upon encounter while reducing CCR5 expression. Upon engraftment into BLT mice, multilineage reconstitution of these ells was observed and viral loads decreased in vivo after HIV challenge [88].
In Vivo Studies on Non-Human Primates
Non-human primates are a good model to understand how certain HIV therapies and stem cell modifications will transplant, reconstitute and perform in a clinical setting. As mentioned frequently in this paper, CCR5 knockdown is a common method of engineering HIV resistance, but the rhesus macaque, a common non-human primate model, has a single nucleotide variation in its CCR5 allele. An et. al, was one of the first to work with sh1005, overcame this hurdle and created a base pair mutation in the sh1005 vector (CCR5-targeting shRNA) to match that of the rhesus macaque and transfected mobilized peripheral blood CD34+ HSPCs [35]. After successful engraftment, they observed multilineage hematopoietic differentiation and a consistent down regulation of rhesus macaque CCR5. They also harvested and selected modified CD4+ T cells and found them to be resistant to simian immunodeficiency virus (SIV) ex vivo [36]. The four rhesus macaque models in this study were studied by Kim et. al. They followed the differentiation and reconstitution of the modified HSPCs in the models and found that modified gene marking remained constant for ten years in these models, despite some variations in the HSPCs [89]. They also found that myeloid and lymphoid cells were normally repopulated from the modified cells and maintained a relatively constant output. These results are significant because they provide evidence that HSPC repopulation can occur with incredible diversity. A later 2009 study by Trobridge et. al focused on HSPCs modified with C46 fusion inhibitor [90]. These cells resisted HIV and SIV infection upon challenge and, to further select for gene-modified cells, were simultaneously transduced with MGMT-P140K marker. This population of modified and resistant CD34+ cells were transplanted into the pigtailed macaque and found modified gene expression in 7% of lymphocytes. They were also able to resist HIV/SIV challenge ex vivo and expressed long-term expression of C46 inhibitor [90]. A second transplantation resulted in a 4-fold increase in modified cell population after in vivo challenge, and since the vectors used in this study are HIV-based, they can be easily transitioned to a clinical study. A 2013 study by Younan et. al also used a C46 inhibitor with MGMT-p140K marker which were transfected into HSPCs and transplanted into pigtail macaques. Using gene marking and SIV challenge, they selected for gene-modified cells and found levels of modified CD4+ T cells over 90% higher than the control group in gastrointestinal tract, lymph nodes, and peripheral blood. They also observed a maintenance of high-frequency gene marking in this group and, interestingly, an enhanced cytotoxic function of T lymphocytes as well as normal function of other antibody responses [91]. They also found that these cells were resistant to in vivo infection of SIV and HIV and decreased viral load. This information is particularly important because it reveals that C46 inhibitor modification does not inhibit the normal function of modified HSPC-derived lymphocytes, and more importantly can mount a response to HIV infection. A 2015 study also from Younan et. al focused on the effects of antiretroviral therapy (ART) and HSPC transplantation, a likely situation for an HIV patient that has already undergone ART [92]. Pigtail macaques were infected with SIV and HIV then treated with ART and were transplanted with CD34+ HSPCs containing C46 inhibitor. This resulted in very low gene marking levels after transplantation and CD34+ cells were measured for ART drugs which was detectable in all and is a possible explanation for low gene transfer. They also noticed high levels of CCR5 expression in CD4+ T cells which suggests that there may need to be an extended period of time between ART and gene modification. Nonetheless, the results can contribute to developing a protocol lentiviral gene modification in later clinical trials for subjects that are receiving ART [92].
Clinical Trials
Clinical trials in anti-HIV HSPC gene therapy began in 1999 where the safety and capabilities of modified HSPC transplantation were evaluated [93]. In the study by Kohn et. al, CD34+ cells from bone marrow of 4 HIV pediatric patients were tested for in vitro transduction using a retroviral vector carrying a decoy gene. These cells were reintroduced to the patients without adverse effect, but gene marking in peripheral blood after one year was less than 0.01% [93]. A phase I trial in 2000 tested 3 different HIV genes. This included a portion of the rev-responsive element (RRE), which is capable of sequestering HIV rev protein, a dominant-negative mutant of tat and rev (HIV regulatory genes), and a hammerhead ribozyme specific to HIV mRNA. CD34+ HSPCs were harvested from 4 pediatric HIV patients’ bone marrow and transduced with the three mentioned anti-HIV genes. The cells were reintroduced to the patients without adverse effects and supported previous evidence that gene modification and transfer to patients is safe. They also found that cells with gene marking showed inhibition of HIV replication as high as 99.5%, but gene marking in was low in peripheral blood after reinfusion (less than 0.01%) [94].
A recent phase II trial by Mitsuyasu et. al tested anti-HIV ribozyme targeting tat/tar (OZ1) and was transduced into CD34+ HSPCs on adult HIV patients [95]. The 74 subjects either received a placebo or OZ1, which was received without adverse events. The goal was to evaluate OZ1 CD34+ HSPCs and their ability to differentiate in vivo into mature lymphoid and myeloid cells that could resist HIV infection and protect from further HIV replication. Over the course of 100 weeks, they found that CD4+ lymphocyte levels were higher in the OZ1 subjects and, more importantly, viral load was consistently lower in this group compared to the control. This was the first study to indicate that gene transfer in cells could be safely introduced to patients and reduce viral load and support differentiation and maturation. An important negative effect from this treatment was seen in mature T lymphocytes. OZ1 subjects saw shorter T lymphocyte lifespan and a reduction in ability to return to marrow [95].
Another clinical trial published in the paper by Digiusto et. al used CD34+ HSPCs and modified them to express 3 anti-HIV genes: CCR5 ribozyme, tat/rev shRNA, and TAR decoy [75]. They transduced the cells via self-inactivating lentiviral vector (SIN) that carried the three anti-HIV genes. Four adult HIV patients were engrafted with the cells and showed no adverse events throughout the 11-day post-infusion evaluation, but gene marking was less than 0.2% for the 24 month trial, thus this treatment did not provide any evidence of benefit [75]. These results reveal the need to design an improved protocol for transduction and transplantation to infuse patients only with modified cells to increase levels of genetic modification.
A 2007 clinical study published in the paper by van Lunzen et. al enrolled 10 HIV-positive adults that were resistant to multidrug treatment and infused them with autologous T lymphocytes that were previously transduced with a retroviral vector expressing C46 inhibitor [60]. No adverse events were reported in any patient and they saw a significant increase in CD4+ cell counts after infusion and remained at or above baseline in the 1-year follow-up. They also detected levels of gene marking in peripheral blood, lymph nodes, and bone marrow throughout the study, but no significant changes in viral load within the first 4 months were observed. However, 7 patients in this trial changed their antiviral drug treatment and 4 of them responded with a significant reduction in viral load [60]. These results are particularly important because they were able to achieve an increase in gene marking in important body systems, but also found that using antiviral drugs in combination with this stem cell treatment proved to be quite effective in reducing viral load. This suggests that a combination of modified stem cell infusion paired with ART might be an effective treatment for patients that do not respond well to or have a resistance to these antiretroviral drugs. The use of C46 inhibitor is a popular approach in engineering HIV immunity in these clinical trials, but there has also been a clinical trial published by Tebas et. al that successfully used CCR5-targeting ZFNs [96]. Autologous CD4+ T cells were modified with this vector and 12 patients were infused with a single dose of these cells to assess the efficacy of this treatment, specifically adverse events, persistence of modified cells, CD4 T cell counts, effects on viral load, and homing to gut mucosa.
One serious adverse event has reported but was associated with a reaction to transfusion. CD4+ T cell counts increased after transfusion, levels of HIV DNA in the blood decreased in all patients and HIV RNA was even undetectable in one patient, which can be explained by the reconstitution of resistant T cells [96]. This evidence shows that ZFN disruption of CCR5 in HSPCs can not only provide resistance to HIV, but also can lead to an increase in cells that do not express CCR5 in HIV-targeting lymphoid and myeloid compartments.
Conclusion
Since the first case of curing HIV through resistant bone marrow transplants, anti-HIV gene therapy has become a promising approach to finding a cure to HIV and is supported by a wide range of studies and methods. There are a multitude of approaches to engineering HIV resistance in HSPCs including ZFN, TALEN, RNAi, and CRISPR/Cas9. They have shown through in vitro and in vivo experiments that these HSPCs (and their progenies) can protect from HIV infection. Most importantly, some of these gene strategies have been investigated in clinical trials which have exemplified the safety and efficacy of engraftment into patients. Because of low engraftment rates and low levels of modified HSPCs, these anti-HIV cells have not reached levels of reconstitution sufficient to serve as an effective therapy. In order for anti-HIV gene therapy to become a permanent cure, modified HSPC engraftment and reconstitution has to be improved. Nonetheless, future studies that achieve significant increases in engraftment could pave the path to solving the issue of life-time HIV immunity without daily drug treatment.
References
- Turner BG, Summers MF. (1999) Structural biology of HIV. J Mol Biol. 285(1):1-32.
- Sundquist WI, Kräusslich HG. (2012) HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med. 2(7):a006924.
- Cao Z, Li J, Chen H, Song C, Shen Z, et al. (2020) Effects of HIV-1 genotype on baseline CD4+ cell count and mortality before and after antiretroviral therapy. Sci Rep. 10(1):15875.
- De Cock KM, Jaffe HW, Curran JW. (2021) Reflections on 40 Years of AIDS. Emerg Infect Dis. 27(6):1553-1560.
- Eggleton JS, Nagalli S. (2021) Highly Active Antiretroviral Therapy (HAART).
- Chung J, Scherer LJ, Gu A, Gardner AM, Torres-Coronado M, et al. (2014) Optimized Lentiviral Vectors for HIV Gene Therapy: Multiplexed Expression of Small RNAs and Inclusion of MGMTP140K Drug Resistance Gene. Mol Ther. 22(5):952-63.
- Sáez-Cirión A, Pancino G. (2013) HIV controllers: a genetically determined or inducible phenotype? Immunol Rev. 254(1):281-94.
- Ramana KV, Sabitha V, Rao R. (2013) A Study of Alternate Biomarkers in HIV Disease and Evaluating their Efficacy in Predicting T CD4+ Cell Counts and Disease Progression in Resource Poor Settings in Highly Active Antiretroviral Therapy (HAART) Era. J Clin Diagn Res. JCDR. 7(7):1332.
- Yang X, Su B, Zhang X, Liu Y, Wu H, Zhang T. (2020) Incomplete immune reconstitution in HIV/AIDS patients on antiretroviral therapy: Challenges of immunological non-responders. J Leukoc Biol. 107(4):597-612.
- Cobucci RN, Lima PH, de Souza PC, Costa VV, Cornetta Mda C, et al. (2015) Assessing the impact of HAART on the incidence of defining and non-defining AIDS cancers among patients with HIV/AIDS: a systematic review. J Infect Public Health. (1):1-10.
- Allers K, Schneider T. (2015) CCR5Δ32 mutation and HIV infection: basis for curative HIV therapy. Curr Opin Virol. 14:24-29.
- Hütter G, Blüthgen C, Neumann M, Reinwald M, Nowak D, et al. (2013) Coregulation of HIV-1 dependency factors in individuals heterozygous to the CCR5-delta32 deletion. AIDS Res Ther. 10(1):1-8.
- Persaud D, Palumbo PE, Ziemniak C, Hughes MD, Alvero CG, et al. (2012) Dynamics of the resting CD4+ T-cell latent HIV reservoir in infants initiating HAART less than 6 months of age. AIDS Lond Engl. 26(12):1483.
- Kordelas L, Verheyen J, Esser S. (2014) Shift of HIV Tropism in Stem-Cell Transplantation with CCR5 Delta32 Mutation. N Engl J Med. 371(9):880-2.
- Henrich TJ, Hanhauser E, Marty FM, Sirignano MN, Keating S, et al. (2014) Antiretroviral-Free HIV-1 Remission and Viral Rebound Following Allogeneic Stem Cell Transplantation: A Report of Two Cases. Ann Intern Med. 161 (5):319-27.
- Hütter G, Bodor J, Ledger S, Boyd M, Millington M, et al. (2015) CCR5 Targeted Cell Therapy for HIV and Prevention of Viral Escape. Viruses. 7(8):4186-203.
- Kiem HP, Jerome KR, Deeks SG, McCune JM. (2012) Hematopoietic-Stem-Cell-Based Gene Therapy for HIV Disease. Cell Stem Cell. 10(2):137-47.
- Glass WG, McDermott DH, Lim JK, Lekhong S, Yu SF, et al. (2006) CCR5 deficiency increases risk of symptomatic West Nile virus infection. J Exp Med. 203(1):35-40.
- Hellier S, Frodsham AJ, Hennig BJW, Klenerman P, Knapp S, et al. (2003) Association of genetic variants of the chemokine receptor CCR5 and its ligands, RANTES and MCP-2, with outcome of HCV infection. Hepatol. 38(6):1468-76.
- Promrat K, McDermott DH, Gonzalez CM, Kleiner DE, Koziol DE, et al. (2003) Associations of chemokine system polymorphisms with clinical outcomes and treatment responses of chronic hepatitis C. Gastroenterol. 124(2):352-60.
- Wald O, Pappo O, Ari ZB, Azzaria E, Wiess ID, et al. (2004) The CCR5Δ32 allele is associated with reduced liver inflammation in hepatitis C virus infection. Eur J Immunogenet. 31(6):249-52.
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, et al. (2013) RNA-guided human genome engineering via Cas9. Science. 339(6121):823-6.
- Cong L, Ran FA, Cox D, Lin S, Barretto R, et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science. 339(6121):819-23.
- Cho SW, Kim S, Kim JM, Kim JS. (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 31(3):230-2.
- Mandal PK, Ferreira LMR, Collins R, Meissner TB, Boutwell CL, et al. (2014) Efficient Ablation of Genes in Human Hematopoietic Stem and Effector Cells using CRISPR/Cas9. Cell Stem Cell. 15(5):643-52.
- Sarver N, Cantin EM, Chang PS, Zaia JA, Ladne PA, et al. (1990) Ribozymes as potential anti-HIV-1 therapeutic agents. Science. 247(4947):1222-5.
- Rossi JJ, June CH, Kohn DB. (2007) Genetic therapies against HIV. Nat. Biotechnol. 25(12):1444-54.
- Cagnon L, Rossi JJ. (2000) Down regulation of the CCR5 β-Chemokine Receptor and Inhibition of HIV-1 Infection by Stable VA1-Ribozyme Chimeric Transcripts. Antisense Nucleic Acid Drug Dev. 10(4):251-61.
- Li MJ, Bauer G, Michienzi A, Yee JK, Lee NS, et al. (2003) Inhibition of HIV-1 Infection by Lentiviral Vectors Expressing pol III-promoted anti-HIV RNAs. Mol Ther. 8(2):196-206.
- Scadden DT, Shen H, Cheng T. (2001) Hematopoietic stem cells in HIV disease. J Natl Cancer Inst Monogr. 2000(28):24-9.
- Panoskaltsis N, Abboud CN. (1999) Human immunodeficiency virus and the hematopoietic repertoire: implications for gene therapy. Front Biosci. 4:D457-67.
- Shen H, Cheng T, Preffer FI, Dombkowski D, Tomasson MH, et al. (1999) Intrinsic human immunodeficiency virus type 1 resistance of hematopoietic stem cells despite coreceptor expression. J Virol. 73(1):728-737.
- Kim DH, Rossi JJ. (2007) Strategies for silencing human disease using RNA interference. Nat Rev Genet. 8(3):173-84.
- Qin X-F, An DS, Chen ISY, Baltimore D. (2003) Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci. 100(1):183-8.
- An DS, Qin FXF, Auyeung VC, Mao SH, Kung SKP, et al. (2006) Optimization and Functional Effects of Stable Short Hairpin RNA Expression in Primary Human Lymphocytes via Lentiviral Vectors. Mol Ther. 14(4):494-504.
- An DS, Donahue RE, Kamata M, Poon B, Metzger M, et al. (2007) Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci. 104(32):13110-5.
- Liang M, Kamata M, Chen KN, Pariente N, An DS, et al. (2010) Inhibition of HIV-1 infection by a unique short hairpin RNA to chemokine receptor 5 delivered into macrophages through hematopoietic progenitor cell transduction. J Gene Med. 12(3):255-65.
- Shimizu S, Hong P, Arumugam B, Pokomo L, Boyer J, et al. (2010) A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood. 115(8):1534-44.
- Shimizu S, Ringpis G E, Marsden MD, Cortado RV, Wilhalme HM, et al. (2015) RNAi-Mediated CCR5 Knockdown Provides HIV-1 Resistance to Memory T Cells in Humanized BLT Mice. Mol Ther Nucleic Acids. 4:e227.
- Zhang HX, Zhang Y, Yin H. (2019) Genome Editing with mRNA Encoding ZFN, TALEN, and Cas9. Mol Ther. 27(4):735-46.
- Mani M, Kandavelou K, Dy FJ, Durai S, Chandrasegaran S. (2005) Design, engineering, and characterization of zinc finger nucleases. Biochem Biophys Res Commun. 335(2):447-57.
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, et al. (2008) Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 26(7):808-16.
- Heyer WD, Ehmsen KT, Liu J. (2010) Regulation of Homologous Recombination in Eukaryotes. Annu Rev Genet. 44:113-39.
- Wang J, Exline CM, DeClercq JJ, Llewellyn GN, Hayward SB, et al. (2015) Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol. 33(12):1256-63.
- Mock U, Machowicz R, Hauber I, Horn S, Abramowski P, et al. (2015) mRNA transfection of a novel TAL effector nuclease (TALEN) facilitates efficient knockout of HIV co-receptor CCR5. Nucleic Acids Res. 43(11):5560-71.
- Mussolino C, Morbitzer R, Lütge F, Dannemann N, Lahaye T, et al. (2011) A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 39(21):9283-93.
- Romito M, Juillerat A, Kok YL, Hildenbeutel M, Rhiel M, et al. (2021) Preclinical Evaluation of a Novel TALEN Targeting CCR5 Confirms Efficacy and Safety in Conferring Resistance to HIV-1 Infection. Biotechnol J. 16(1):e2000023.
- Gaj T, Gersbach CA, Barbas CF. (2013) ZFN, TALEN and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 31:397-405.
- Cradick TJ, Fine EJ, Antico CJ, Bao G. (2013) CRISPR/Cas9 systems targeting β-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 41:9584.
- Ye L, Wang J, Beyer AI, Teque F, Cradick TJ, et al. (2014) Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Δ32 mutation confers resistance to HIV infection. Proc Natl Acad Sci USA. 111(26):9591-6.
- Kleinstiver BP, Pattanayak V, Prew MS, Tsai SQ, Nguyen NTet al. (2016) High-fidelity CRISPR–Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 529:490-95.
- Zhang C, Zhu R, Cao Q, Yang X, Huang Z, et al. (2020) Discoveries and developments of CXCR4-targeted HIV-1 entry inhibitors. Exp Biol Med (Maywood). 245(5):477-85.
- Anderson J, Banerjea A, Planelles V, Akkina R. (2003) Potent Suppression of HIV Type 1 Infection By a Short Hairpin Anti-CXCR4 siRNA. AIDS Res. Hum. Retroviruses. 19:699-706.
- Yuan J, Wang J, Crain K, Fearns C, Kim KA, et al. (2012) Zinc-finger Nuclease Editing of Human cxcr4 Promotes HIV-1 CD4+ T Cell Resistance and Enrichment. Mol Ther. 20:849-59.
- Schumann K, Lin S, Boyer E, Simeonov DR, Subramaniam M, et al. (2015) Generation of knock-in primary human T cells using Cas9 ribonucleoproteins. Proc Natl Acad Sci. 112:10437-42.
- Bianchi ME, Mezzapelle R. (2020) The Chemokine Receptor CXCR4 in Cell Proliferation and Tissue Regeneration. Front Immunol. 11:2109.
- Hildinger M, Dittmar MT, Schult-Dietrich P, Fehse B, Schnierle BS, et al. (2001) Membrane-Anchored Peptide Inhibits Human Immunodeficiency Virus Entry. J Virol. 75:3038-42.
- Zahn RC, Hermann FG, Kim E-Y, Rett MD, Wolinsky SM, et al. (2008) Efficient Entry Inhibition of Human and Non-Human Primate Immunodeficiency Virus by Cell Surface-Expressed gp41-Derived Peptides. Gene Ther. 15:1210-222.
- Burke BP, Levin BR, Zhang J, Sahakyan A, Boyer J, et al. (2015) Engineering Cellular Resistance to HIV-1 Infection In Vivo Using a Dual Therapeutic Lentiviral Vector. Mol Ther. 4:e236.
- van Lunzen J, Glaunsinger T, Stahmer I, von Baehr V, Baum C, et al. (2007) Transfer of Autologous Gene-modified T Cells in HIV-infected Patients with Advanced Immunodeficiency and Drug-resistant Virus. Mol Ther. 15:1024-33.
- Wolstein O, Boyd M, Millington M, Impey H, Boyer J, et al. (2014) Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol Ther Methods Clin Dev. 1:11.
- Chan E, Towers GJ, Qasim W. (2014) Gene Therapy Strategies to Exploit TRIM Derived Restriction Factors against HIV-1. Viruses. 6:243-63.
- Yap MW, Nisole S, Stoye JP. (2005) A single amino acid change in the SPRY domain of human Trim5alpha leads to HIV-1 restriction. Curr Biol. 15:73-8.
- Neagu MR, Ziegler P, Pertel T, Strambio-De-Castillia C, Grütter C, et al. (2009) Potent inhibition of HIV-1 by TRIM5-cyclophilin fusion proteins engineered from human components. J Clin Invest. 119:3035-47.
- Karpinski J, Hauber I, Chemnitz J, Schäfer C, Paszkowski-Rogacz M, et al. (2016) Directed evolution of a recombinase that excises the provirus of most HIV-1 primary isolates with high specificity. Nat. Biotechnol. 34:401-09.
- Qu X, Wang P, Ding D, Li L, Wang H, Ma L, et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013;41:7771–7782.
- Ebina H, Misawa N, Kanemura Y, Koyanagi Y. (2013) Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci Rep. 3:2510.
- Wang G, Zhao N, Berkhout B, Das AT. (2016) CRISPR-Cas9 Can Inhibit HIV-1 Replication but NHEJ Repair Facilitates Virus Escape. Mol Ther. 24:522-26.
- Tiley LS, Brown PH, Cullen BR. (1990) Does the human immunodeficiency virus Tat trans-activator contain a discrete activation domain? Virology. 178(2):560-7.
- Lee M-TM, Coburn GA, McClure MO, Cullen BR. (2003) Inhibition of Human Immunodeficiency Virus Type 1 Replication in Primary Macrophages by Using Tat- or CCR5-Specific Small Interfering RNAs Expressed from a Lentivirus Vector. J Virol. 77:11964.
- Strong CL, Guerra HP, Mathew KR, Roy N, Simpson LR, et al. (2015) Damaging the Integrated HIV Proviral DNA with TALENs. PLOS ONE. 10:e0125652.
- ter Brake O, ’t Hooft K, Liu YP, Centlivre M, Jasmijn von Eije K, et al. (2008) Lentiviral Vector Design for Multiple shRNA Expression and Durable HIV-1 Inhibition. Mol Ther. 16:557-64.
- Walker JE, Chen RX, McGee J, Nacey C, Pollard RB, et al. (2012) Generation of an HIV-1-Resistant Immune System with CD34+ Hematopoietic Stem Cells Transduced with a Triple-Combination Anti-HIV Lentiviral Vector. J Virol. 86:5719-29.
- Anderson J, Li M-J, Palmer B, Remling L, Li S, et al. (2007) Safety and Efficacy of a Lentiviral Vector Containing Three Anti-HIV Genes—CCR5 Ribozyme, Tat-rev siRNA, and TAR Decoy—in SCID-hu Mouse–Derived T Cells. Mol Ther. 15:1182-88.
- DiGiusto DL, Krishnan A, Li L, Li H, Li S, et al. (2010) RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Sci Transl Med. 2:36ra43.
- Ringpis G-EE, Shimizu S, Arokium H, Camba-Colón J, Carroll MV, et al. (2012) Engineering HIV-1-Resistant T-Cells from Short-Hairpin RNA-Expressing Hematopoietic Stem/Progenitor Cells in Humanized BLT Mice. PLOS ONE. 7:e53492.
- Wolstein O, Boyd M, Millington M, Impey H, Boyer J, et al. (2014) Preclinical safety and efficacy of an anti-HIV-1 lentiviral vector containing a short hairpin RNA to CCR5 and the C46 fusion inhibitor. Mol Ther Methods Clin Dev. 1:11.
- Shultz LD, Brehm MA, Garcia-Martinez JV, Greiner DL. (2012) Humanized mice for immune system investigation: progress, promise and challenges. Nat Rev Immunol. 12(11):786-98.
- Holt N, Wang J, Kim K, Friedman G, Wang X, et al. (2010) Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 28:839-47.
- Saydaminova K, Ye X, Wang H, Richter M, Ho M, et al. (2015) Efficient genome editing in hematopoietic stem cells with helper-dependent Ad5/35 vectors expressing site-specific endonucleases under microRNA regulation. Mol Ther Methods Clin Dev. 1:14057.
- Hofer U, Henley JE, Exline CM, Mulhern O, Lopez E, et al. (2013) Pre-clinical Modeling of CCR5 Knockout in Human Hematopoietic Stem Cells by Zinc Finger Nucleases Using Humanized Mice. J Infect Dis. 208:S160-S164.
- Li L, Krymskaya L, Wang J, Henley J, Rao A, Cao L-F, et al. (2013) Genomic Editing of the HIV-1 Coreceptor CCR5 in Adult Hematopoietic Stem and Progenitor Cells Using Zinc Finger Nucleases. Mol Ther. 21:1259-69.
- Li M-J, Kim J, Li S, Zaia J, Yee J-K, et al. (2005) Long-Term Inhibition of HIV-1 Infection in Primary Hematopoietic Cells by Lentiviral Vector Delivery of a Triple Combination of Anti-HIV shRNA, Anti-CCR5 Ribozyme, and a Nucleolar-Localizing TAR Decoy. Mol Ther. 12:900-09.
- Myburgh R, Ivic S, Pepper MS, Gers-Huber G, Li D, et al. (2015) Lentivector Knockdown of CCR5 in Hematopoietic Stem and Progenitor Cells Confers Functional and Persistent HIV-1 Resistance in Humanized Mice. J Virol. 89:6761-772.
- Centlivre M, Legrand N, Klamer S, Liu YP, von Eije KJ, et al. (2013) Preclinical In Vivo Evaluation of the Safety of a Multi-shRNA-based Gene Therapy Against HIV-1. Mol Ther. 2:e120.
- Kalomoiris S, Lawson J, Chen RX, Bauer G, Nolta JA, et al. (2012) CD25 Preselective Anti-HIV Vectors for Improved HIV Gene Therapy. Hum. Gene Ther Methods. 23:366-75.
- Melkus MW, Estes JD, Padgett-Thomas A, Gatlin J, Denton PW, et al. (2006) Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat Med. 12:1316-22.
- Zhen A, Kamata M, Rezek V, Rick J, Levin B, et al. (2015) HIV-specific Immunity Derived From Chimeric Antigen Receptor-engineered Stem Cells. Mol Ther. 23:1358-67.
- Kim S, Kim N, Presson AP, Metzger ME, Bonifacino AC, et al. (2014) Dynamics of HSPC Repopulation in Nonhuman Primates Revealed by a Decade-Long Clonal-Tracking Study. Cell Stem Cell. 14:473-85.
- Trobridge GD, Wu RA, Beard BC, Chiu SY, Muñoz NM, et al. (2009) Protection of Stem Cell-Derived Lymphocytes in a Primate AIDS Gene Therapy Model after In Vivo Selection. PLoS ONE.
- Younan PM, Polacino P, Kowalski JP, Peterson CW, Maurice NJ, et al. (2013) Positive selection of mC46-expressing CD4+ T cells and maintenance of virus specific immunity in a primate AIDS model. Blood. 122:179-87.
- Younan PM, Peterson CW, Polacino P, Kowalski JP, Obenza W, et al. (2015) Lentivirus-mediated Gene Transfer in Hematopoietic Stem Cells Is Impaired in SHIV-infected, ART-treated Nonhuman Primates. Mol Ther J Am Soc Gene Ther. 23:943-51.
- Kohn DB, Bauer G, Rice CR, Rothschild JC, Carbonaro DA, et al. (1999) A Clinical Trial of Retroviral-Mediated Transfer of arev-Responsive Element Decoy Gene Into CD34+ Cells From the Bone Marrow of Human Immunodeficiency Virus-1–Infected Children. Blood. 94:368-71.
- Bauer G, Selander D, Engel B, Carbonaro D, Csik S, et al. (2000) Gene Therapy for Pediatric AIDS. Ann N Y Acad Sci. 918:318-29.
- Mitsuyasu RT, Merigan TC, Carr A, Zack JA, Winters MA, et al. (2009) Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat Med. 15:285-92.
- Tebas P, Stein D, Tang WW, Frank I, Wang SQ, et al. (2014) Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N Engl J Med. 370:901-10.
Genesis Scientific Publication is licensed under CC BY-NC-ND 4.0![]()
![]()
![]()
![]()
