Restoration of GABAA Receptor Function after Benzodiazepine Use: A Meta-Analysis
Halford G Warlick, Gabriel N De Souza and Vincent S Gallicchio*
Department of Biological Sciences, College of Science, Clemson University, Clemson, USA
*Corresponding author: Vincent S Gallicchio, Department of Biological Sciences, College of Science, Clemson University, Clemson, USA
Citation: Warlick HG, De Souza GN, Gallicchio VS. (2020) Restoration of GABAA Receptor Function after Benzodiazepine Use: A Meta-Analysis. J Stem Cell Res. 1(1):1-19.
Received Date: June 25, 2020, | Published Date: July 09, 2020
Copyright© 2020 genesis pub by Warlick HG, et al. CC BY-NC-ND 4.0 DEED. This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-No Derivatives 4.0 International License., This allows others distribute, remix, tweak, and build upon the work, even commercially, as long as they credit the authors for the original creation.
DOI: https://doi.org/10.52793/JSCR.2020.1(1)-05
Abstract
Benzodiazepines (BZDs) are some of the most commonly prescribed medications in the United States and are currently classified as schedule IV-controlled substances. Originally patented in 1958, Librium became the first BZD synthesized and set the stage for what would become a major change in medical practice. Since their introduction, BZDs were meant to replace barbiturates, a precursor family with highly addictive properties and lethal overdose consequences. Within the past twenty years, it has been found that BZDs can also cause physical dependence, sometimes in as little as two weeks, and addiction because of their strong relaxant and euphoric properties. Research has hypothesized that dependency occurs because of GABAA receptor desensitization and functional down regulation. This meta-analysis compiles multiple literature sources to determine novel ways to restore GABAA receptor function so that recovering patients can maintain long-term abstinence from BZDs. Selected psychiatric medications and their pharmacology are discussed to determine potential combinations that may normalize GABAA receptor activity after chronic BZD use.
Keywords
Benzodiazepines; Receptor; Drugs; Stress disorder; Psychiatric medications; Pharmacology; Spinal cord; Treatment; Central nervous system.
Abbreviations
AMPA: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BAR: Barbiturate; BZD: Benzodiazepine; CBD: Cannabidiol; CBZ: Carbamazepine; DZP: Diazepam; GABA: χ -aminobutyric acid; MML: Muscimol; NMDA: N-methyl-D-aspartate; CNS: Central Nervous System
Introduction
Gamma-(γ−-aminobutyric acid (GABA) is the primary inhibitory neurotransmitter in the mammalian brain. GABA is highly important in homeostatic regulation because it reduces neural excitability, regulates mood, and modulates inhibitory neurotransmission [1]. GABA has two receptors in the central nervous system (CNS) with distinctive functions: GABAA and GABAB [1]. The GABAA receptor is an ionotropic receptor, and the GABAB receptor is a G-protein coupled receptor [2]. Ionotropic receptors undergo a conformational change and open when agonists are present, allowing ion passage into intracellular fluid [3]. G-protein coupled receptors induce cellular responses by using guanine-exchange nucleotide factors, cyclic AMP, and adenylate cyclase within the transmembrane domain [4].
In clinical practice, both receptors are targets for drugs. Baclofen binds to the GABAB receptor to induce myorelaxation, and BZDs and alcohol attach to the GABAA receptor to enhance anxiolysis and promote sleep [5]. The GABAA receptor (Figure 1) is a pentameric heterooligomer that consists of five subunits (2α, 2β, 1γ) surrounding a ligand-gated chloride channel.

Figure 1: GABAA Receptor.
The Cl-channel is enclosed by two GABA receptors, binding one GABA molecule each, and a BZD receptor, binding one BZD molecule. Each GABA binding site is located between α and β subunits, and the BZD binding site is found between α and γ subunits [8]. Six Isoforms of the alpha subunit exist, but only four of these are BZDs sensitive (α1, α2, α3, α5) [9]. The most common Isoforms that comprise the BZD receptor are the α1 and α2 subunits. α1 receptors are widely distributed in the brain and are typically found in the cerebellum, cortex and thalamus and are responsible for the sedative, amnesic, and anticonvulsant effects of BZDs [10]. (Alpha-2 (α2) receptors are involved with anxiolys and myorelaxation and tend to be more localized, predominating in the limbic system, spinal cord, and motor neurons [11]. Since each isoform is involved with specific effects, a small fraction of BZDs favor one Isoform over the other. Temazepam, a hypnotic BZD, favors the α1 Isoform and has strong hypnotic properties [12]. Temazepam also has anxiolytic effects albeit less than the nonselective BZDs [13].
Long-Term Effects of Benzodiazepines
BZDs are widely prescribed to treat insomnia, panic disorder, post-traumatic stress disorder, muscle spasms, and epilepsy [14]. Physicians tend to prescribe BZDs for extended periods of time, sometimes over the course of years. However, the Food and Drug Administration (FDA) warns that severe psychological and/or physical dependency may develop in almost a third of individuals taking BZDs consistently over a month [15].
Case-reports and documentaries have revealed that patients taking BZDs long-term are at high risk for severe withdrawal symptoms. According to a Lisa Ling CNN documentary, a patient reported having to micro-taper clonazepam in order to avoid treatment reinstatement. Micro-tapering is not used in traditional medicine and has solely been developed by patients trying to come off BZDs. This should alert physicians to change their prescribing habits. Chronic BZD use also produces unwanted side-effects such as cognitive decline, memory impairment, and motor retardation [16]. One observational study reported that individuals treated with diazepam over a two-year period had significant gray matter reduction in the motor cortex and reduced processing speed [17].
Pharmacokinetics
Pharmacokinetics refers to how the body processes a drug [18]. Each medication has its own pharmacokinetic profile that depends on its administration site and hepatic metabolism [18]. Sublingual, intramuscular, and intravenous administration of BZDs does not bypass the liver and retains a high bioavailability. Oral formulations are required to undergo hepatic metabolism before being released into the bloodstream, reducing their bioavailability. This process is known as the first-pass effect [18].
During the first-pass effect in the liver, BZDs undergo their first stage of metabolism before entering the bloodstream. The BZD is broken down into metabolites, and the active metabolites are then released into the circulation. The significance of the first-pass effect depends on the specific BZD [18]. Alprazolam retains a favorable pharmacokinetic profile because its bioavailability decreases by only 20% after metabolism [19]. On the contrary, midazolam’s bioavailability is drastically reduced by 50% due to extensive hepatic metabolism [20]. It must be noted that not all BZDs undergo hepatic metabolism. Some are metabolized through the kidneys like oxazepam, which may benefit liver-impaired patients.
BZDs also vary significantly in their lipophilic properties. BZDs that have favorable lipophilic structures are easily absorbed by the small intestine and reach the bloodstream faster than less lipophilic BZDs [21]. Interestingly, the lipophilic structure of the BZD determines its amnesic effects [22,23]. Alprazolam (Figure 2), a highly lipophilic BZD, induces stronger amnesic effects than clonazepam (Figure 3), a poorly lipophilic BZD [24, 25]. Clonazepam possesses a positively charged amino group on its diazepine backbone (Figure 4), giving it a significantly higher charge than alprazolam, explaining its reduced amnesic effects. These phenomena illustrate the importance of pharmacokinetics and how drug structure determines its pharmacodynamic properties.

Figure 2: Alprazolam.

Figure 3: Clonazepam.

Figure 4: Benzodiazepine Structure.
BZDs also break down into their own distinctive metabolite(s). Diazepam metabolizes into nordiazepam, temazepam, and oxazepam before exiting the body [26]. Clonazepam breaks down into 7-amino-clonazepam and is only detectable in the body as such [26]. Since metabolites from long-acting BZDs are stored in the body long after excretion, residual inactive metabolites may still induce unwanted side effects. As a result, the elderly should not be prescribed long-acting BZDs like diazepam because of increased fall risk, confusion, and cognitive dysfunction [27].
Modulation of GABAA Receptor Function
BZDs act as positive allosteric modulators (PAMs) at the GABAA receptor by increasing GABA binding affinity [28]. Multiple theories have been proposed about how GABA efficacy increases after BZD binding. The most commonly accepted notion states that BZDs increase GABA affinity by inducing a conformational change within the GABAA receptor [7]. A newer theory posits that BZDs cause receptor equilibrium to shift from the closed state to the opened state, indirectly increasing GABA binding affinity [29].
The first notion mentions that BZDs simply increase GABAA’s affinity for GABA by inducing a conformational change within the receptor [7]. GABAA’s extracellular domain and transmembrane domain must interact favorably for this structural change to occur. When the conformation does change, GABA can bind more easily and induce larger inhibitory currents than normal. In support of this hypothesis, one case-study found that specific residues within the transmembrane domain were required to allow subunit-receptor gating [29]. A chimeric study expanded on this theory and found that BZDs changed the γ2 subunit’s conformation within the transmembrane domain. This implies that the γ2 subunit is responsible for GABAA’s conformational change. Interestingly, this research found that negative allosteric modulators did not alter γ2 activity, suggesting that γ2 activity is mediated exclusively by PAMs.
Another model states that the GABAA receptor exists in three different conformations, each with a distinctive opened and closed state. These conformations include the non-liganded state, the mono-liganded state, and the di-liganded state. The non-liganded state does not have a GABA molecule attached to it, while the mono-liganded and di-liganded states have one and two GABA molecules bound to them respectively [31]. Proponents of this model suggest that BZDs shift receptor equilibrium from the closed state to the opened state, increasing GABA binding probability [31]. The probability model directly challenges the traditional conformational theory. However, neither model has proven to be entirely accurate nor the actual modulation mechanism remains to be discovered.
Tolerance Mechanisms
Tolerance develops rapidly over the course of months after chronic BZD treatment and is dependent on the drug’s half-life [32]. Half-life is the time that it takes for half of a drug to be metabolized and removed from the bloodstream by renal excretion [32]. Long-acting BZDs (e.g. clonazepam, diazepam, chlordiazepoxide) have half-lives between 40-200 hours while short acting BZDs (e.g. alprazolam, lorazepam) have half-lives between 8-25 hours [33]. Consequently, tolerance develops faster with the shorter acting BZDs. At the molecular level, the mechanism of tolerance is best understood by observing each subunit’s structural and/or functional change after BZD treatment. In addition to the subunit’s efficacy, one must also understand GABAA’s mode of desensitization.
Several experiments suggest that the α1 subunit loses its effectiveness over extended periods of BZD exposure, resulting in reduced hypnotic, anticonvulsive, and sedative effects [8]. Multiple observational studies have found that after six weeks, sleep-deprived patients began to experience rebound insomnia and required a dose increase in order to maintain sleep [11]. On the contrary, the α2 subunit’s efficacy remains unchanged after long-term BZD use, allowing BZDs to retain their anxiolytic and myorelaxant effects [34]. Neither theory has been proven, but consistent data from multiple studies has given us insight into the neuropharmacology of these drugs.
Receptor desensitization models have been disputed between scientists for decades. One theory posits that GABAA receptor numbers decrease after chronic BZD treatment due to intracellular lysosomal degradation [34]. A recent experiment challenged this model and found that GABAA receptor numbers remained unchanged after chronic diazepam (DZP) treatment in mice [35]. Since receptor numbers did not decrease after DZP treatment, DZP did not reduce receptor count [35].
Newer theories suggest that receptor uncoupling and/or NMDA (N-methyl-D-aspartate)/AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) upregulation may be responsible for tolerance [36]. The uncoupling hypothesis mentions that the GABAA receptor’s subunits become uncoupled over time [8]. Effectively, the “link” between the GABA binding site and BZD binding site becomes dysfunctional, requiring more BZD to be present in order to maintain the BZD-GABA subunit interaction. Scientists in favor of this hypothesis suggest that allosteric modulation efficacy decreases over time, inducing tolerance [37]. The data is inconclusive, however, and further research is needed to formulate a clear assumption. The NMDA/AMPA model posits that NMDA and AMPA receptors become upregulated after BZD use. Since NMDA and AMPA receptors are excitatory and respond to glutamate, upregulation of these receptors increases excitatory activity. This may precipitate withdrawal symptoms and cause seizures, involuntary movements, and tremors [38]. One study endorsing this hypothesis reports that DZP treatment with an NMDA antagonist inhibited tolerance in mice [39]. A second experiment found that NMDA receptor numbers were upregulated after DZP treatment. Controversially, other research found that AMPA receptor numbers remained unchanged after DZP treatment, refuting the NMDA/AMPA hypothesis [10].
Medications that may Restore GABAA Receptor Function Gabapentinoids
Gabapentinoids (Figure 5) include gabapentin and pregabalin [40]. These GABA-mimetic compounds possess anti-nociceptive, anti-convulsant, and anxiolytic properties [41]. Pregabalin and gabapentin act similarly in the brain, but pregabalin is absorbed more readily by the small intestine and has a higher bioavailability than gabapentin. The gabapentinoids do not interact or affect the GABAA receptor in any way. They primarily exert their effects by reducing voltage-gated calcium channel activity in the axon terminal that effectively inhibits glutamate release. Gabapentinoids modulate GABA metabolism by increasing GABA synthesis and release into the extra synaptic space. Because the excess GABA is found in the extra synaptic space and not in the inter-neural synaptic space, it has no effects on the GABAA receptor [42].

Figure 5: Gabapentinoid.
Phenibut
Phenibut (Figure 6) is an anxiolytic and non-tropic compound that originated from Russia in the 1960’s [43]. Phenibut is available by prescription in most countries but sold online in the United States. Phenibut has a distinctive mechanism of action from other anxiolytic compounds because it is a GABA derivative and it agonizes both the GABAA and GABAB receptors [44].

Figure 6: Phenibut.
It is able to rapidly cross the BBB due to its phenyl ring and exerts its effects within only two hours of absorption [45]. Phenibut promotes cognitive functioning and has direct dopaminergic effects, unlike the BZDs. It is associated, however, with moderate dependency and BZD-like withdrawals if taken in large quantities [46].
Kava Lactones
The kava lactones (Figure 7), derived from the Piper methysticum plant, have been used in traditional medicine for thousands of years because of their anxiolytic and hypnotic effects [47,48]. Research suggests that these compounds block voltage-gated sodium channels, enhance GABA binding and reduce glutamate release. Kava lactones have also been shown to inhibit both norepinephrine reuptake and monoamine oxidase B activity, while also suppressing eicosanoid thromboxane synthesis. Their mechanism of action is similar to that of the BZDs, but the kava lactones do not have tolerance or dependency risks [48].

Figure 7: Kava Lactone.
Flumazenil
Flumazenil (Figure 8) was developed and approved in 1991 for BZD overdose. Flumazenil is only available in intravenous (IV) form at hospitals and requires careful monitoring [49]. This drug acts as a BZD binding antagonist and displaces BZDs from their receptor, reversing sedation. Flumazenil has also been shown to upregulate GABAA receptor function in vitro, providing promising results. However, flumazenil can greatly decrease seizure threshold and induce delirium/psychosis at high doses [50].

Figure 8: Flumazenil.
Cannabidiol
Cannabidiol (CBD) (Figure 9) is a non-psychotropic agent derived from the Cannabis sativa plant [51]. CBD possesses antipsychotic and anxiolytic properties that may deliver marked psychiatric benefits. Evidence has shown that CBD plays a major role in GABAA receptor modulation by β subunit binding. When attached to the GABAA receptor, CBD amplifies GABA-evoked currents much like the BZDs. Studies have suggested that CBD may treat some dependent patients but may also induce and/or worsen psychosis in patients with mental health disorders [52].

Figure 9:Cannabidiol.
Barbiturates
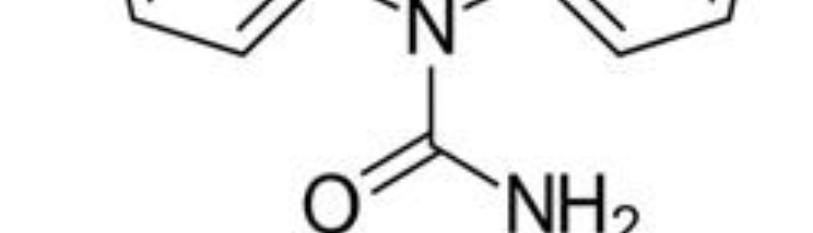
Barbiturates (BARs) (Figure 10) were first synthesized in 1864 and treated anxiety and sleep disorders for almost a century [53]. BARs bind to the GABAA receptor and alter its function much like BZDs. They modulate GABAA activity by increasing the chloride channel opening duration, which produces significantly larger inhibitory currents than the BZDs. BARs also block excitatory AMPA and kainite receptors, giving them powerful depressive properties. As a result, BARs have a significantly lower therapeutic index than BZDs and can easily cause overdose by respiratory depression. BARs may profoundly affect GABAA receptor function like BZDs and cause receptor uncoupling. Withdrawal symptoms tend to be more severe than the BZDs because of the marked effects on CNS depression [54].

Figure 10: Barbiturate.
Carbamazepine
Carbamazepine (CBZ) (Figure 11) is an anticonvulsant medication used to treat seizures, nerve pain, and bipolar disorder [55]. CBZ interacts with the GABAA receptor by increasing inhibitory chloride current in GABAergic neurons. Studies have reported that CBZ modulates GABAA receptorlike the BZDs but remains unaffected by flumazenil. CBZ has been extensively studied for BZD dependence and may play a role in GABAA receptor modulation. However, its side-effect profile is unfavorable and limits its use because of potential blood dyscrasia and liver toxicity [56].

Figure 11: Carbamazepine.
Sodium Valproate
Sodium valproate (Figure 12) is a medication used to treat epilepsy and bipolar disorder [57]. Sodium valproate blocks voltage-gated sodium channel activity and increases synaptic GABA levels by inhibiting GABA degradative enzymes. It has been used in clinical trials for BZD dependence, but has serious side-effects and constant blood monitoring is often required to prevent liver toxicity. Research into its effects on GABAA receptor function has been limited but suggests that the increased GABA levels may contribute to receptor normalization.

Figure 12: Sodium valproate.
Jujubogenin
Jujubogenin (Figure 13) is a sapogenin extracted from the Ziziphus jujube seeds and has a relatively high affinity for the GABAA receptor [59]. During its early use thousands of years ago, jujubogenin was found to possess anxiolytic properties due to bioactive ingredients like the flavonoids and phenolics [60]. Recent evidence has suggested that it also retains neuroprotective properties and can rapidly penetrate the BBB. Because of its high solubility and easy passage into the brain, it exerts marked therapeutic effects and may promote GABAA receptor normalization [61].

Figure 13: Jujubogenin.
Muscimol
Muscimol (MML) (Figure 14) is a psychoactive compound derived from the mushroom Amanita muscaria [62]. MML exhibits depressant, sedative-hypnotic and hallucinogenic activity after purification. MML acts as a GABAA agonist by selectively binding to the δ subunit on the GABA binding site, distinguishing it from most other GABAA PAMs. Muscimol demonstrates excellent therapeutic effects but must be monitored closely in humans due to lack of research and potential risks in patients withdrawing from BZD [63].

Figure 14: Muscimol.
Discussion
According to Table 1, each medication has its own unique pharmacotherapeutic profile. Our literature search revealed that most selected medications interacted directly with the GABAA receptor and modulated its activity similarly. The gabapentinoids (Figure 5) were the only medications that did not interact with the GABAA receptor. They were found to have a marked effect on GABA metabolism and extrasynaptic GABA levels and may prove useful in increasing GABA availability [41]. The antiepileptic drugs carbamazepine (CBZ) (Figure 11) and sodium valproate (Figure 12) were found to act almost identically, except that sodium valproate increased GABA levels, while CBZ did not [64,65]. The increased GABA levels and GABAA modulation may make sodium valproate a better candidate for receptor normalization than CBZ. Phenibut (Figure 6) was found to interact with both GABA receptor subtypes and agonize both the ligand-gated chloride channel in the GABAA receptor and guanine-exchange protein system in the GABAB receptor. Phenibut may be a good option in restoring receptor function in combination with other compounds. Multiple literature studies reported that both flumazenil (Figure 8) and barbiturates (Figure 10) are high-risk medications because flumazenil can induce seizures, and barbiturates can cause respiratory depression [66,67]. These medications serve little to no purpose in receptor restoration, except when flumazenil is administered for a significant amount of time [66]. The naturalistic compounds we analyzed had comparable pharmacodynamic properties tour pharmaceutical drugs, most notably kava lactones (Figure 7). The reason for this lies in evolutionary biology and conservation of receptor proteins for thousands of years. Regardless of naturalistic or synthetic origin, each drug retains its own side effect profile and must be thoroughly analyzed before BZD trials occur. Combinations of medications with similar pharmacodynamic properties may prove useful in restoring the conformation of the GABAA receptor, but their detailed pharmacological interactions are beyond the scope of this literature review.
|
Medication |
Modulation |
Clinical Use |
GABAA Modulation |
|
Gabapentinoids |
GABA synthesis modulator; Ca2+ channel blocker |
Antinociceptive; anticonvulsant |
Anxiolysis; increased GABA may help restore GABAA receptor function |
|
Phenibut |
GABAA and GABAB receptor PAM |
Anxiolytic |
BZD withdrawal; restores GABAA receptor function |
|
Kava Lactones |
GABAA receptor PAM |
Anxiolytic; sedative |
BZD withdrawal |
|
Flumazenil |
BZD receptor antagonist |
BZD overdose |
BZD withdrawal; GABAA receptor upregulation |
|
Cannabidiol |
GABAA receptor PAM |
Antidepressant; antinociceptive |
GABAA receptor coupling |
|
Barbiturates |
GABAA receptor PAM |
Anxiolytic; sedative |
BZD withdrawal; gates Cl- channel |
|
Carbamazepine |
Blocks Na+ channel; modulates Cl- channel |
Anticonvulsant; mood stabilizer |
Restores GABAA receptor inhibitory action |
|
Valproate |
Na+ channel blocker; increases GABA levels |
Anticonvulsant; mood stabilizer |
Restores GABAA receptor conformation |
|
Jujubogenin |
Increases GABA levels |
Anxiolytic; hypnotic |
Increases GABA levels; restores GABAA receptor function |
|
Muscimol |
Positively modulates GABAA receptor function |
Sedative-hypnotic; hallucinogen |
Restores GABAA receptor modulation |
Table 1: Pharmacodynamics of GABAA Modulators.
Two potential medications that may restore GABAA receptor function are the gabapentinoids (Figure 5) and muscimol (MML) (Figure 14). Since gabapentinoids increase extrasynaptic GABA levels and block Ca2+ channel activity, more GABA is available to bind to extrasynaptic receptors. Because gabapentinoids also block excitatory Ca2+ channel activity, they inhibit glutamate release and prevent NMDA/AMPA receptor activation. Since NMDA/AMPA receptors become upregulated during BZD withdrawal, less glutamate prevents this upregulation [42]. Therefore, the gabapentinoids may serve an important role in BZD dependence. MML also exhibits promising properties. Research has shown that MML is potent GABAA agonist and binds directly to the δ subunit on the GABA binding site [63]. Stimulating the δ subunit is unique for MML since none of our other medications activated this isoform. In addition, MML is not an allosteric modulator and does not induce tolerance like the BZDs, yet interestingly produces similar effects [63]. If gabapentinoids and MML were used together, their pharmacology may benefit GABAA conformation normalization. Since MML is a GABAA agonist and gabapentinoids increase extrasynaptic GABA levels, their combined actions may alleviate withdrawal symptoms. Increased GABA concentrations and a GABAA agonist may stimulate the GABAA receptor enough to allow its gating structure to return to its normal state. Thus, our hypothesis supports the NMDA/AMPA and GABAA uncoupling models and rejects the receptor down regulation theory.
A second possible medication combination that may promote GABAA receptor restoration includes the kava lactones (Figure 7) and carbamazepine (Figure 11). Kava lactones act on the GABAA receptor similarly to BZDs, and CBZ increases GABAA inhibitory currents [48,56]. Neither kava lactones nor CBZ are affected by flumazenil, suggesting they are uninvolved with tolerance or dependency [68]. Because CBZ increases inhibitory Cl- channel conductance, it may attenuate GABAA membrane potential during BZD withdrawal [64]. Kava lactones enhance GABAA binding and prevent NMDA/AMPA receptor upregulation, while suppressing glutamate release [68]. In addition, these compounds block voltage-gated sodium channel activity and inhibit norepinephrine reuptake [68]. Neither of these drugs has been approved by the FDA for sedative-hypnotic withdrawal, but they may have potential benefit in restoring receptor function. Since kava lactones act much like BZDs and CBZ promotes inhibitory currents in the neuron, their combined effects may agonize receptor function, while also ensuring the resting membrane potential remains at or below -70 mV [68]. However, proper dosing, medical oversight, and regular blood work are required for CBZ because CBZ may disrupt red blood cell production. Kava lactones have little to no side effects but may interact with other medications [64,68].
Evidence on flumazenil (Figure 8) treatment reports low GABAA modulation and limited therapeutic benefit. Some case-studies have reported improved withdrawal symptoms after flumazenil treatment but have been limited. Major drawbacks of flumazenil treatment include required IV use and decreased seizure threshold. Parenteral injection is required because oral tablets and capsules are unavailable. Medical supervision is also necessary because electrolyte balance and fluid levels may become disrupted during treatment [69]. Flumazenil may also interfere with other medications and reduce their effectiveness. Affordability and insurance coverage are also limited and may present financial problems for certain patients. Thus, flumazenil’s side-effects, required medical supervision, and cost limit its therapeutic value.
Previous studies have shown that CBD (Figure 9) and phenibut (Figure 6) positively modulate GABAA receptor activity. At low doses, these compounds may be effective in treating BZD dependence. Case-studies have reported a notable increase in α1 and α2 receptor expression following CBD treatment and α1 subunit expression after phenibut administration [44,52]. These models support CBD’s anticonvulsant and antinociceptive properties, as well as phenibut’s anxiolytic effects [43,52]. Much like the gabapentinoids, CBD decreases intracellular calcium levels and neuronal excitability. Because of this, CBD treatment may stabilize NMDA and AMPA receptor levels. One experiment used cannabidivarin (a CBD derivative) with GABAergic neurons and found it helped couple GABAA receptor subunits together after an epileptic fit [52]. Since GABAA receptor uncoupling is directly caused by subunit phosphorylation and dephosphorylation, this research supports the uncoupling hypothesis [8]. It does not, however, support the receptor down regulation model because cannabidivarin was not found to affect GABAergic receptor numbers. In regard to phenibut, multiple case-studies demonstrate that it increases presynaptic GABA release and inhibits voltage-dependent calcium channel activity after α2/γ subunit binding. This decreases excitatory receptor activity and promotes relaxation and anxiolysis [43-44]. Nevertheless, research is lacking into phenibut’s effects on NMDA/AMPA activity and thus provides limited insight into any other GABAA receptor model. However, phenibut may be habit-forming and produce withdrawal symptoms in as little as 2 weeks if abused [70]. One observational study reported a patient who had abused phenibut and had to be treated with a six-week long phenobarbital taper [70]. Thus, attempting to restore GABAA receptor function may prove difficult for phenibut because of addiction liability. Interestingly, phenibut may have clinical benefit when combined with CBD because it potentiates CBD’s depressant properties. Since CBD stabilizes NMDA/AMPA receptors and phenibut inhibits glutamate release, the NMDA/AMPA receptor model is most likely supported with these two drugs.
Even though barbiturates (Figure 10) are effective in treating epilepsy, anxiety, and insomnia, they have a markedly low therapeutic index compared to BZDs. BARs are PAMs at the GABAA receptor site and alter anion influx by increasing Cl- channel opening duration, rather than frequency like the BZDs. BARs block AMPA and kainite receptors, agonize GABA binding at high doses, and suppress glutamate release [71]. Their strong effects on CNS activity may cause life-threatening respiratory depression, bradycardia, and induced coma. Because of this, they have little to no benefit on GABAA receptor normalization and may actually further dysregulate GABAA function. The only relevant use of BARs involves acute treatment of BZD withdrawal if a BZD was unavailable for tapering. Long-term use of BARs to restore GABAA receptor function is impractical.
Future Research
Even though some aspects of BZD pharmacology have been understood by scientists over the past few decades, there is much to discover. GABAA receptor function is still poorly grasped, and research with humans has been limited due to potential harmful drug effects. Experiments with mice models have shown that chronic BZD use leads to tolerance and certain subunits may be involved with this. Therefore, restoration of GABAA receptor function needs to be clearly understood before performing human experiments. If this is clarified, scientists can develop several experiments to investigate the GABAA receptor and ways to alter its conformation back to normal. This would revolutionize the field of psychopharmacology.
Since BZD withdrawal leads to a host of symptoms such as tremors, muscle spasms, seizures, insomnia, and rebound anxiety, providing our selected medications to patients and evaluating their symptoms may benefit our current knowledge of the GABAA receptor. Selecting the correct medication(s) and providing the right dose could be challenging, and adverse reactions may have severe consequences if incorrectly monitored. Each individual is unique and responds differently to medications than others. Perhaps creating an experiment that compares drug responses between a control group (non-BZD dependent) and treatment group (BZD-dependent) could be of interest for future research. As a hypothetical example, if a control group received CBZ and the treatment group received CBZ and there was symptom relief within the treatment group, CBZ may have an effect on GABAA restoration.
Another potential experiment could involve positron-emission topography imaging of the brain between control subjects and treatment subjects. The imaging would compare glucose metabolism between the two groups prior to and after taking our medication. If the treatment group’s glucose metabolism shifted to appear more like the control’s after receiving the drug, that medication may have a potential benefit. Imaging the actual GABAA receptor on human neurons is beyond the scope of current medical research; however, monitoring patients’ response to medications and glucose uptake may elucidate GABAA receptor pharmacology and allow scientists to implement these drugs into detoxification facilities to assist patients with getting off BZDs.
Conclusion
Since their discovery over 60 years ago, BZDs are renowned medications in medical practice. These drugs are extremely helpful in treating insomnia, anxiety, neurological disorders, and agitation in the short-term. Chronic use is never recommended because of severe dependency and/or addiction. Using synthetic or naturalistic compounds to restore GABAA receptor function may be helpful in dependent patients. The medications we analyzed through our literature search all had some effect on the GABA and/or the GABAA receptor and implementing them into clinical practice may be the first step in understanding GABAA receptor dysfunction. Research is needed in this area because millions of Americans take these medications long-term. Without understanding receptor dysfunction and medication treatment, BZD-dependent individuals will continue to suffer undesirable withdrawal symptoms.
Acknowledgments
We greatly thank each and every research institution for developing these experiments and observational studies.
Conflict of Interest
The authors have no conflict of interest at this time.
References
- Fluyau D, Revadigar N, Manobianco BE. (2018) Challenges of the pharmacological management of benzodiazepine withdrawal, dependence, and discontinuation. Ther Adv Psychopharmacol. 8(5):147-168.
- Möhler H. (2002) Pathophysiological aspects of diversity in neuronal inhibition: a new benzodiazepine pharmacology. Dialogues Clin Neurosci. 4(3):261-9.
- Vinkers CH, Olivier B. (2012) Mechanisms Underlying Tolerance after Long-Term Benzodiazepine Use: A Future for Subtype-Selective GABA(A) Receptor Modulators? Adv Pharmacol Sci. 2012:416864.
- Kacirova I, Grundmann M, Silhan P, Brozmanova H. (2016) A Case Report of Clonazepam Depen;dence: Utilization of Therapeutic Drug Monitoring During Withdrawal Period. Medicine. 95(9):e2881.
- Ahuja T, Mgbako O, Katzman C, Grossman A. (2018) Phenibut (β-Phenyl-γ-aminobutyric Acid) Dependence and Management of Withdrawal: Emerging Nootropics of Abuse. Hindawi.
- Jacob TC, Michels G, Silayeva L, Haydon J, Succol F, et al. (2012) Benzodiazepine treatment induces subtype-specific changes in GABA(A) receptor trafficking and decreases synaptic inhibition. Proc Natl Acad Sci USA. 109(45):18595-600.
- Vinkers CH, Olivier B. (2012) Mechanisms Underlying Tolerance after Long-Term Benzodiazepine Use: A Future for Subtype-Selective Receptor Modulators? Adv Pharmacol Sci. 2012:1-19.
- Griffin CE, Kaye AM, Bueno FR, Kaye AD. (2013) Benzodiazepine pharmacology and central nervous system-mediated effects. Ochsner J. 13(2):214-23.
- Susman J, Klee B. (2005) The Role of High-Potency Benzodiazepines in the Treatment of Panic Disorder. Prim Care Companion J Clin Psychiatry. 7(1):5-11.
- Tan KR, Rudolph U, Lüsche, C. (2011) Hooked on benzodiazepines: GABAA receptor subtypes and addiction. Trends Neurosci. 34(4):188-197.
- Bergmann R, Kongsbak K, Sørensen PL, Sander T, Balle T. (2013) A unified model of the GABA(A) receptor comprising agonist and benzodiazepine binding sites. PloS one. 8(1):e52323.
- Reynolds LM, Engin E, Tantillo G, Lau HM, Muschamp JW, et al. (2012) Differential roles of GABA(A) receptor subtypes in benzodiazepine-induced enhancement of brain-stimulation reward. Neuropsychopharmacolog. 37(11):2531-40.
- Schmitz A. (2016) Benzodiazepine use, misuse, and abuse: A review. Ment Health Clin. 6(3):120-6.
- Guina J, Merrill B. (2018) Benzodiazepines I: Upping the Care on Downers: The Evidence of Risks, Benefits and Alternatives. J Clin Med. 7(2):17.
- Cheng T, Wallace DM, Ponteri B, Tuli M. (2018) Valium without dependence? Individual GABAA receptor subtype contribution toward benzodiazepine addiction, tolerance, and therapeutic effects. Neuropsychiatr Dis Treat. 14:1351-61.
- Vinitskiĭ IM, Dubrovina NI, Il'iuchenok RIu. (1985) Vliianie diazepama na protsessy interferential [Effect of diazepam on interference processes]. Farmakol Toksikol. 48(4):38-41.
- Sadock BJ, Sadock VA, Ruiz P, Kaplan HI. (2015) Kaplan & Sadocks synopsis of psychiatry: Behavioral sciences, clinical psychiatry. Philadelphia: Wolters Kluwer.
- Scavone JM, Greenblatt DJ, Shader RI. (1987) Alprazolam kinetics following sublingual and oral administration. J Clin Psychopharmacol. 7(5):332-34.
- Paine MF, Shen DD, Kunze KL, Perkins JD, Marsh CL, et al. (1996) First-pass metabolism of midazolam by the human intestine. Clin Pharmacol Ther. 60(1):14-24.
- Bergmann R, Kongsbak K, Sørensen PL, Sander T, Balle T. (2013) A unified model of the GABA(A) receptor comprising agonist and benzodiazepine binding sites. PloS one. 8(1):e52323.
- Maria G. (2016). Effect of Chronic Benzodiazepine Exposure on GABAA Receptors. Neuropathol Drug Addict Subst Misuse. 3:308-16
- National Center for Biotechnology Information. PubChem Database. Benzodiazepine, CID=134664.
- National Center for Biotechnology Information. PubChem Database. Alprazolam, CID=2118.
- National Center for Biotechnology Information. PubChem Database. Clonazepam, CID=2802.
- Huang W, Moody DE. (1995) Immunoassay detection of benzodiazepines and benzodiazepine metabolites in blood. J Anal Toxicol. 19(6):333-342.
- Mangoni AA, Jackson SH. (2004) Age-related changes in pharmacokinetics and pharmacodynamics: basic principles and practical applications. Br J Clin Pharmacol. 57(1):6-14.
- Zhu S, Noviello CM, Teng J, Walsh RM, Jr Kim, et al. (2018) Structure of a human synaptic GABAA receptor. Nature. 559(7712):67-72.
- Gielen MC, Lumb MJ, Smart TG. (2012) Benzodiazepines modulate GABAA receptors by regulating the preactivation step after GABA binding. J. Neurosci. 32:5707-571.
- Hanson SM, Czajkowski C. (2008) Structural mechanisms underlying benzodiazepine modulation of the GABA(A) receptor. J Neurosci. 28(13):3490-99.
- Campo-Soria C, Chang Y, Weiss DS. (2006) Mechanism of action of benzodiazepines on GABAA receptors. Br J Pharmacol. 148(7):984-90.
- Hallfors DD, Saxe L. (1993) The dependence potential of short half-life benzodiazepines: a meta-analysis. Am J Public Health. 83(9):1300-04.
- Burrows GD, Norman TR, Judd FK, Marriott PF. (1990) Short-acting versus long-acting benzodiazepines: discontinuation effects in panic disorders. J Psychiatr Res. 24 Suppl, 2:65-72.
- Chang Y, Ghansah E, Chen Y, Ye J, Weiss DS. (2002) Desensitization mechanism of GABA receptors revealed by single oocyte binding and receptor function. J Neurosci. 22(18):7982-90.
- Steppuhn KG, Turski L. (1993) Diazepam dependence prevented by glutamate antagonists. Proc Natl Acad Sci USA. 90(14):6889-893.
- Prevot TD, Li G, Vidojevic A, Misquitta KA, Fee C, et al. (2019) Novel Benzodiazepine-Like Ligands with Various Anxiolytic, Antidepressant, or Pro-Cognitive Profiles. Mol Neuropsychiatry. 5(2):84-97.
- McGoldrick MK, Galanopoulou AS. (2014) Developmental pharmacology of benzodiazepines under normal and pathological conditions. Epileptic disorders. 16(Spec No 1):S59-S68.
- Iacobucci GJ, Popescu GK. (2017) NMDA receptors: linking physiological output to biophysical operation. Nat Rev Neurosci. 18:236.
- Petrus DS, Fabel K, Kronenberg G, Winter C, Steiner B, et al. (2009) NMDA and benzodiazepine receptors have synergistic and antagonistic effects on precursor cells in adult hippocampal neurogenesis. Eur J Neurosci. 29(2):244-52.
- National Center for Biotechnology Information. PubChem Database. Gabapentin, CID=3446.
- Elena P Calandre, Fernando Rico-Villademoros, Mahmoud Slim. (2016) Alpha2delta ligands, gabapentin, pregabalin and mirogabalin: a review of their clinical pharmacology and therapeutic use. Expert Rev Neurother. 16(11):1263-77.
- Schifano F. (2014) Misuse and abuse of pregabalin and gabapentin: cause for concern? CNS drugs. 28(6):491-6.
- National Center for Biotechnology Information. PubChem Database. 4-Amino-3-phenylbutanoic acid, CID=14113.
- Hardman MI, Sprung J, Weingarten TN. (2019) Acute phenibut withdrawal: A comprehensive literature review and illustrative case report. Bosn J Basic Med Sci. 19(2):125-9.
- Sivakumar Nathasha AP, Anitha R, Ganapathy D. (2019) Therapeutic benefits of phenibut-A review. Drug Invent Today. 11:2012-16.
- McCabe DJ, Bangh SA, Arens AM, Cole JB. (2019)Phenibut exposures and clinical effects reported to a regional poison center. Am J Emerg Med. 37(11):2066-071.
- National Center for Biotechnology Information. PubChem Database. Kavaform, CID=6444199.
- Singh YN, Singh NN. (2002) Therapeutic potential of kava in the treatment of anxiety disorders. CNS Drugs. 16(11):731-43.
- National Center for Biotechnology Information. PubChem Database. Flumazenil, CID=3373.
- Whitwam JG, Amrein R. (1995) Pharmacology of flumazenil. Acta Anaesthesiol Scand Suppl. 108:3-14.
- National Center for Biotechnology Information. PubChem Database. Cannabinol, CID=2543.
- Cifelli P, Ruffolo G, De Felice E, Alfano V, van Vliet EA, et al. (2020) Phytocannabinoids in Neurological Diseases: Could They Restore a Physiological GABAergic Transmission? Int J Mol Sci. 21(3):723.
- National Center for Biotechnology Information. PubChem Database. Barbituric acid, CID=6211.
- Skibiski J, Abdijadid S. Barbiturates. (2019) In: Stat Pearls Treasure Island (FL): Stat Pearls Publishing.
- National Center for Biotechnology Information. PubChem Database. Carbamazepine, CID=2554.
- Tolou-Ghamari Z, Zare M, Habibabadi JM, Najafi MR. (2013) A quick review of carbamazepine pharmacokinetics in epilepsy from 1953 to 2012. J Res Med Sci. 18(Suppl 1):S81-5.
- National Center for Biotechnology Information. PubChem Database. Valproic acid, CID=3121.
- Baldino F Jr, Geller HM. (1981) Sodium valproate enhancement of gamma-aminobutyric acid (GABA) inhibition: electrophysiological evidence for anticonvulsant activity. J Pharmacol Exp Ther. 217(2):445-50.
- National Center for Biotechnology Information. PubChem Database. Jujuboside B, CID=24721031.
- Manayi A, Nabavi SM, Daglia M, Jafari S. (2016) Natural terpenoids as a promising source for modulation of GABAergic system and treatment of neurological diseases. Pharmacol Rep. 68(4):671-79.
- You ZL, Xia Q, Liang FR, Tang YJ, Xu CL, et al. (2010) Effects on the expression of GABAA receptor subunits by jujuboside A treatment in rat hippocampal neurons. J Ethnopharmacol.128(2):419-23.
- National Center for Biotechnology Information. PubChem Database. Muscimol, CID=4266.
- Benkherouf AY, Taina KR, Meera P, Aalto AJ, Li XG, et al. (2019). Extrasynaptic δ-GABAA receptors are high-affinity muscimol receptors. J Neurochem. 149(1):41-53.
- Macdonald RL, Kelly KM. (1995) Antiepileptic drug mechanisms of action. Epilepsia. 36Suppl 2:S2-S12.
- Löscher W, Schmidt D. (1980) Increase of human plasma GABA by sodium valproate. Epilepsia. 21(6):611-15.
- An H, Godwin J. (2016) Flumazenil in benzodiazepine overdose. CMAJ. 188(17-18):E537.
- López-Muñoz F, Ucha-Udabe R, Alamo C. (2005) The history of barbiturates a century after their clinical introduction. Neuropsychiatr Dis Treat. 1(4):329-43.
- Tzeng YM, Lee MJ. (2015) Neuroprotective properties of kavalactones. Neural Regen Res. 10(6):875-77.
- Hood SD, Norman A, Hince DA, Melichar JK, Hulse GK. (2014) Benzodiazepine dependence and its treatment with low dose flumazenil. Br J Clin Pharmacol. 77(2):285-94.
- Brunner E, Levy R. (2017) Case Report of Physiologic Phenibut Dependence Treated With a Phenobarbital Taper in a Patient Being Treated With Buprenorphine. J Addict Med. 11(3), 239-40.
- Suddock JT, Cain MD. (2020) Barbiturate Toxicity. In: StatPearls Treasure Island (FL): Stat Pearls Publishing.
- Skibiski J, Abdijadid S. Barbiturates (2019). In: Stat Pearls.
Genesis Scientific Publication is licensed under CC BY-NC-ND 4.0![]()
![]()
![]()
![]()
