Identification of Novel Frameshift Variant in PUS1Gene by Whole Exome Sequencing
Romina Maleknia1,2, Farid Momeni1,2, Firooze Ronassian1,2, Mohamad Javad Maleknia1,2, Sajad Rafiee Komachali3*, and Mansoor Salehi1,2,4
1Cellular, Molecular, and Genetics Research Center, Isfahan University of Medical Sciences, Isfahan, Iran
2Medical Genetics Research Center of Genome, Isfahan University of Medical Sciences, Isfahan, Iran
3Department of Biology, University of Sistan and Baluchestan, Zahedan 98167-45845, Iran
4Department of Medical Genetics, School of Medicine, Isfahan University of Medical Sciences, Isfahan, Iran
*Corresponding author: Sajad Rafiee Komachali, Department of Biology, University of Sistan and Baluchestan, Zahedan, Iran.
Citation: Maleknia R, Momeni F, Ronassian F, Maleknia MJ, Komachali SF, et al. (2023) Identification of novel frameshift variant in PUS1 gene by whole exome sequencing.Genesis J Surg Med.2(1):1-06.
Received: January 02, 2023 | Published: January 17, 2023
Copyright© 2023 genesis pub by Malekina F, et al. CC BY-NC-ND 4.0 DEED. This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-No Derivatives 4.0 International License. This allows others distribute, remix, tweak, and build upon the work, even commercially, as long as they credit the authors for the original creation.
DOI: https://doi.org/10.52793/GJSM.2023.2(1)-11
Abstract
Introduction
Myopathy, lactic acidosis, and sideroblastic anemia 1 (MLASA1) is an autosomal recessive inheritance and a mitochondrial disorder associated with the PUS1 gene. It is also associated with bone marrow and skeletal muscle and bone; its related phenotypes are high arc palate and myopathy. This study describes a 30-year-old woman with an MLASA1 phenotype.
Clinical report:
The patient was diagnosed with developmental delay, progressive gait abnormality, and proximal and distal weakness in the lower limb. She also showed symptoms of bilateral facial paresia, lordosis, scapular winging, anemia, colored skin rash, and mild cognitive problems.
Methodology:
Whole exome sequencing identified a novel variant in PUS1. Mutation c.365delG was then confirmed by PCR and sanger sequencing. The patient’s parents’ segregation analysis established the homozygosity and autosomal recessive inheritance pattern for c.365delG.
Results and Calculation:
This study discusses the relationship between this novel mutation in PUS1 and MLASA1 phenotype and its inheritance pattern in a consanguineous family.
Keywords
MLASA; PUS1; Lactic acidosis sideroblastic; Anemia
Introduction
Myopathy, lactic acidosis, and sideroblastic anemia (MLASA) is a mitochondrial respiratory chain disorder (MRCD) associated with mutations in PUS1, YARS2, and MTATP6. Exercise intolerance, muscle weakness, Micrognathia, Hypotonia, High arc palate, and Distichiasis, bone marrow, and skeletal muscle and bone are frequent symptoms of this disease. Delayed puberty, Glaucoma, Intellectual disability, and Microcephaly are among the less frequent ones [1]. MLASA1 (600462# OMIM) is associated with mutations in PUS1 gene, and MLASA2 (613561#OMIM) and MLASA3 (500011#OMIM) are respectively associated with mutations in the YARS2 gene (NM_001040436.2) at 12p11.21 and MTATP6 [2]. PUS1 encodes the nuclear pseudouridine synthase 1, which pseudouridinylates tRNA. Pseudouridine in tRNAs is involved in stopping codon suppression, translational specificity, and recoding efficiency [3]. Mutation in this gene could confront patients lacking pseudouridine in their tRNAs. Pseudo uridine affects the structure of tRNAs by affecting base- stacking in the anticodon loop. When it manifests in stems or the anticodon, it strengthens base-pairing. The stabilized conformation of the anticodon stem-loop could help to keep correct anticodon-codon pairings during translation. This stability improves translation accuracy by slowing down the rate of peptide bond formation, allowing incorrect codon-anticodon pairs to be discarded more frequently.[6] Table 3 shows all variants of PUS1 presented in HGMD. Mutation c.365de. is a likely pathogenic, homozygous variant with an autosomal recessive inheritance pattern located at chromosome 12 exon 3. (p.S123Qfs*12). This variant is a novel mutation that has not been reported to the HGMD, gnomAD, and clinVar databases. This study reports a novel mutation in PUS1, found by whole exome sequencing and convinced by PCR and sanger sequencing, and discusses the relation of this mutation with MLASA type1 (600462# OMIM) based on clinical reports and analyses.
Methods
The patient’s whole exome sequencing (WES) data, with a coverage depth of 150x, was obtained from the patient’s genomic DNA (gDNA). The gDNA was isolated from the patient’s blood specimen using a filter-based methodology. After quality control and quantification, the DNA sample was prepared, and the Sequencing libraries were generated using Agilent Sure Select Human All ExonV7 kit. In the next step, 180- 280bp fragments were generated by aydro dynamic shearing system, modified, and enriched in a PCR reaction. After purifying and quantifying the products, the sequences were generated using NovaSeq 6000 Illumina sequencers. The following steps were performed on a Unix- based operating system. Fast QC and Illu QC tools were used for QC and trimming the sequencing data. Burrows-Wheeler Alignment Tool (BWA) was used for aligning the sequence reads to the GRCH37/UCSC hg19 human reference Genome. Latterly, Picard and GATK packages were applied for base quality score recalibration (BQSR). Then, variant calling and VQSR filtering were applied, and for the last step, the VCF file was annotated using ANNO- VAR, and the results were filtered to find the candidate variants. After checking all the remaining variants in OMIM, the variants were classified based on the American College of Medical Genetics (ACMG) and Association for Molecular Pathology criteria (AMP) protocols. After choosing the candidate variant, DNA extraction, PCR, and Sanger sequencing were performed to con- firm the validation of the selected mutation. The patient’s genomic DNA was extracted using in- nuPREP Blood DNA Mini Kit anger, and Sanger sequencing was operated using an ABI prism 3730 sequencer.
Results
Clinical Report
Patient
This study involves a 30-year-old woman with consanguine parents who presented with progressive gait abnormality and proximal and distal weakness in the lower limb, followed by upper limbs from 6 years ago. She could not walk or crawl until 3 years old, and from 6 years ago, her muscular weakness grew. She cannot walk without help, has difficulty going upstairs, and currently uses a wheelchair. She has weak hands and only lifts lightweight objects. She has a positive Gower sign and mild MR. She had bilateral facial paresia, lordosis, scapular winging, anemia, colored skin rash, and mild cognitive problems and received blood at 4 months old. She had developmental delays. She does not have myalgia, cramps, osteoporosis, or cardiopulmonary. The paradigm tree of the proband has been represented in Figure 1.

Figure 1: Pedigree of the family.
Test and Results
Proband’s brain MR study with multi-planner images (MRI) in different pulse sequences shows normal results. BMA and BMB reveal mild erythroid hyperplasia with no evidence of fibrosis. The muscle biopsy is within normal. Table4 shows the patient’s blood test reports after receiving blood. R.B.C level is lower, and the M.C.V and E.S.R factors and LDL levels are higher than the normal range. CPK levels is 33.5 and LDH is 26.8.EMG and NCV tests diagnosed myopathy Table 1.
|
Test Result Unit |
Reference Interval |
|
F.B.S 81 mg\dL |
60 − 110 |
|
creatinine 0.6 mg\dL |
F: 0.6 − 1.3 |
|
S.G.O.T(AST) 15 IU/L |
up to 32 |
|
S.G.P.T(ALT) 16 IU/L |
F :< 32 |
|
Alkaline Phosphatase 116 IU/L |
61 − 306 |
|
L.D.H 238 U/L |
< 180 |
|
Bilirubin total 0.9 mg\dL |
0.2 − 1.2 |
|
Bilirubin Direct 0.1 mg\dL |
0 − 0.3 |
|
Bilirubin indirect 0.80 mg\dL |
|
|
C.B.C |
|
|
W.B.C 3890 µl |
4000 − 10000 |
|
R.B.C 3.19 x106\µL |
4.25 − 5.4 |
|
Hemoglobin 10.5 g/dL |
12.3 − 15.5 |
|
Hematocite 32.8 % |
36 − 45 |
|
M.C.V 102.8 f L |
80 − 98 |
|
M.C.H 32.9 pg |
27 − 32 |
|
M.C.H.C 32.0 g/dL |
33 − 38 |
|
R.D.W 21.9 % |
11.6 − 14.6 |
|
M.P.V 9.6 f L |
- |
|
Platelets 252000 µl |
150000 − 450000 |
|
E.S.R 1 hrs 20 mm/h |
Up to 15 |
Table 1: Patient’s BloodTest.
Mutation Analysis
After performing the whole exome sequencing analysis that was previously elucidated in section.2,194857 annotated variants were identified. The annotated file was again filtered indifferent stages, and a myopathy panel was applied to initiate the most compatible variants with the proband’s symptoms. Lastly, the homozygous variants were selected due to the con-sanguineous relationship of the patient’s parents, so the number of filtered variants was lessened to a total of 21. The number of selected variants after each filtering step can be seen in Figure 2.

Figure 2: Variant filtering workflow.
All the variants were checked on OMIM and classified via ACMG/AMP protocols. This novel mutation (NM_025215: exon3:c.365delG:p.S123Qfs*12) was marked as likely pathogenic on the Var Somes databases. Because this mutation of the PUS1 gene has a frameshift, it is marked as PVS1. Also, because it has not been reported to gnom AD, ithasaPM2labelaswell.This newly found mutation, located in exon 3 of PUS1, was then confirmed using PCR and sanger sequencing. To produce a 591-based-pair segment for PCR, the forward and Reverse primer sequences, GATTCCATTCCA-CACTACCCACC and AAGCAAAGACCAGAGAGAACACAG, were designed by the Oligo7software Table 2. The PCR steps are as follows: (1) denaturation at 95°C for 5 minutes Table 2.
|
|
Sequence (5′−> 3′) |
GenerunnerTm |
CG% |
|
Forward primer |
GATTCCATTCCACACTACCCACC |
60.94 |
52.17 |
|
Reverse primer |
AAGCAAAGACCAGAGAGAACACAG |
60.98 |
45.83 |
|
product length |
591bp |
- |
- |
Table 2: Primer pairs designed by the Oligo7 software.
(2) amplification by using 30 cycles at 95°C for30 seconds, 60°C for 30 seconds, and 72°C for 30 seconds.
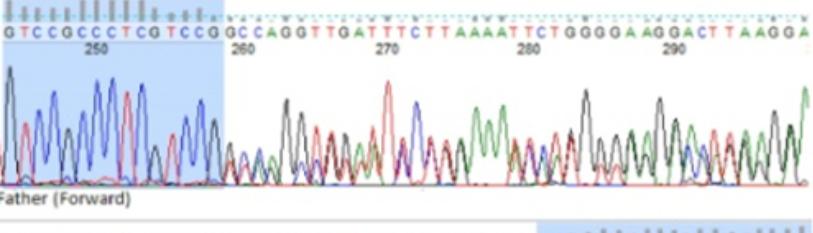
(3) Extension at 72°C for 5 minutes. The PCR’s steps and protocols are represented in Figure 3. Finch TV was used for analyzing the DNA sequences. Figure 2-Figure 4 shows the patient’s parents selected forward and reverse sequences (GTCCGCC-CTCGTCCG and TCAGGCTGTATTCCTGAAA), which confirms the homozygosity for c. 365delG. Thus, it was concluded that this mutation has an auto somal recessive inheritance pattern.


Discussion
The MLASA is caused by the absence of significant reduction of tRNA pseudouridylation. Pseudouridylation is a posttranscriptional modification and the most abundant modification in cellular RNA. Some of its roles in the cell are translation, localization, and stabilization of RNA. Transcription alters a variety of chemically distinct modifications of the RNA [9]. Pseudouridine is vital in anticodon stem-loop (ASL) and stabilizes the ASL’s dynamic structure. There- fore, it makes the proper binding of tRNAs to the ribosome [10]. PUS1 Converts specific uridines to PSI in several tRNA substrates and acts on positions 27/28 in the anticodon stem and positions 34 and 36 in the anticodon of an intron containing tRNA [11]. Pseudouridine causes anticodon codon pairings during translation to go well, increasing translational accuracy in general [12].
From this article, it can be concluded that the novel mutation of PUS1(p.Ser123GlnfsTer12) is associated with myopathy, lactic acidosis, and sideroblastic anemia 1 (MLASA1). This report discussed the case of a proband, a 30-year-old woman diagnosed with myopathy, progressive gait abnormality, proximal and distal weakness in the lower and upper limbs, and developmental delays. After performing WES, a novel mutation of the PUS1 gene (p.Ser123GlnfsTer12) was found on her genomic DNA. This gene encodes a Pseudouridine synthesis involved in the Pseudouridylation process. This study is the first report of this novel mutation in the PUS1 gene, and it shows that thePUS1 is responsible for MLASA1, and this result is compatible with the tests and the patient’s clinical reports.
Authors Contribution
Data curation: Sajad Rafiee Komachali. Clinical report analysis: Farid Momeni, Mohamad Javad Maleknia, Firooze Ronassian; Methodolgy: Romina Maleknia, Sajad Rafiee Komachali; Writing original draft: Romina Maleknia, Farid Momeni; Writing review & editing: Romina Maleknia, Mohamad Javad Maleknia, Sajad Rafiee Komachali; Funding acquisition: Mansoor Salehi.
Acknowledgement
The authors would like to thank the patient and their parents for participating and the Genetics Research Center of Genome for supporting this study.
Ethical Approval
All the patients gave their informed consent for inclusion in this research.
References
Genesis Scientific Publication is licensed under CC BY-NC-ND 4.0![]()
![]()
![]()
![]()
